Болезнь Фридрейха
Семейная атаксия Фридрейха – наследственное дегенеративное заболевание нервной системы, характеризующееся синдромом поражения задних и боковых канатиков спинного мозга. Тип наследования аутосомно-рецессивный, с неполной пенетрантностью патологического гена. Мужчины и женщины болеют одинаково часто.
Что провоцирует / Причины Болезни Фридрейха:
Патогенез (что происходит?) во время Болезни Фридрейха:
Обнаруживаются дегенеративные изменения в проводящих путях задних и боковых канатиков спинного мозга, преимущественно пучков Голля, в меньшей степени – Бурдаха, Флексига, Говерса, волокнах пирамидного пути, задних корешках, а также в клетках коры мозжечка, подкорковых ганглиев, коры большого мозга.
Симптомы Болезни Фридрейха:
Среди экстраневральных проявлений болезни Фридрейха необходимо выделить поражение сердца, которое, по нашим данным, встречается более чем у 90 % больных. Характерным является развитие типичной прогрессирующей кардиомиопатии. Кардиомиопатия преимущественно носит характер гипертрофической, но в отдельных случаях, как следует из наших наблюдений, возможно развитие дилатационной кардиомиопатии. Не исключено, что эти изменения сердца при болезни Фридрейха являются различными стадиями одного процесса. Кардиомиопатия проявляется болями в области сердца, сердцебиением, одышкой при физической нагрузке, систолическим шумом и другими симптомами. Более чем у половины больных кардиомиопатия является непосредственной причиной смерти. Соответствующие изменения обычно обнаруживаются на ЭКГ (нарушение ритма, инверсия зубца Т, изменения проводимости) и при эхокардиографии. В ряде случаев клинические и электрокардиографические симптомы поражения сердца иногда на несколько лет опережают появление неврологических нарушений. Больные длительно наблюдаются у кардиолога или участкового терапевта, чаще всего с диагнозом “ревмокардит”.
Другим характерным экстраневральным проявлением болезни Фридрейха являются скелетные деформации: сколиоз, “стопа Фридрейха” (высокий вогнутый свод стопы с переразгибанием пальцев в основных фалангах и сгибанием в дистальных), деформация пальцев рук и ног и др. Эти нарушения также могут появляться задолго до развития первых неврологических симптомов.
Болезнь Фридрейха характеризуется неуклонно прогрессирующим течением, длительность болезни обычно не превышает 20 лет. Непосредственными причинами смерти могут быть сердечная и легочная недостаточность, инфекционные осложнения.
Диагностика Болезни Фридрейха:
Информативными для диагностики болезни Фридрейха являются электрофизиологические исследования. Характерный для данного заболевания электронейромиографический паттерн заключается в отсутствии или значительном снижении амплитуды потенциалов действия чувствительных нервов конечностей, при сравнительно небольшом снижении скорости проведения импульса по двигательным нервам. Даже в начальной стадии болезни Фридрейха необходимо применять электрокардиографию и эхокардиографию, исследовать содержание глюкозы в крови с проведением специальных нагрузочных тестов толерантности к глюкозе (для исключения сахарного диабета), а также проводить рентгеновское исследование позвоночника (характеристика костных деформаций).
Лечение Болезни Фридрейха:
Применяются симптоматические средства: общеукрепляющие препараты, лечебная физкультура, массаж. В некоторых случаях производится хирургическая коррекция деформации стоп.
К каким докторам следует обращаться если у Вас Болезнь Фридрейха:
Наследственная атаксия фридрейха что это
а) Атаксия Фридрейха. Атаксия Фридрейха — наиболее четко описанная и часто встречающаяся спиноцеребеллярная дегенерация. Частота встречаемости гена составляет 1:110 человек в Англии (Harding 1981a), и примерно один из 10000 человек в Швеции имеет клинические проявления.
Ген атаксии Фридрейха включает повторы ГА А последовательности в интроне 1, который распространен у пациентов (120-1700 повторов). Продуктом нормального гена является белок фратаксин, функция которого не полностью ясна. 94% пациентов с типичной атаксией Фридрейха являются гомозиготами по ГАА экспансии, тем не менее продолжительность повтора на каждой хромосоме из пары неодинакова (Durr et al., 1996a).
В редких случаях отмечается только одна мутация, но в такой ситуации выявляется точечная мутация в гомозиготном локусе (Campuzano et al., 1996). Выраженная длина повтора коррелирует с началом в раннем возрасте, более стремительным течением и наличием кардиомиопатии (Durr et al., 1996a).
Второй ген на хромосоме 9p23-p11 является причиной редких случаев (Фридрейха 2), клинически нечетко отличаемых от 1 типа (Christodoulou et al., 2001).
Основным патологическим проявлением является дистальная аксональная нейропатия, которая поражает нейроны длинных восходящих и нисходящих трактов спинного мозга и крупные сенсорные волокна периферических нервов и ганглии задних корешков (Said et al., 1986). Также зарегистрирована утрата нервных волокон в зрительных путях, а мозжечок остается непораженным.
Сердце увеличено, и более чем в половине случаев отмечается гипертрофическая кардиомиопатия с некрозом волокон и фиброзом, преимущественно затрагивающим левый желудочек.
Критерии диагностики атаксии Фридрейха (Harding, 1981a) включают начало до 25 лет (обычно до 16 лет), аутосомно-рецессивное наследование и сочетанное поражение крупных сенсорных волокон периферических нервов, мозжечкового тракта, пирамидного тракта и задних столбов.
Тем не менее, степень фенотипической вариабельности велика, в некоторых случаях отмечается позднее начало и/или меньшая выраженность симптомов и вариабельное течение, и некоторые пациенты прикованы к инвалидной коляске в раннем подростковом возрасте, в то время как другие способны самостоятельно передвигаться почти до 40 лет (Montermini et al., 1997).
По неофициальным данным, к доминантным случаям относится большая часть наследственной моторной и сенсорной нейропатии со скелетными деформациями и утратой чувствительности, но некоторые случаи не поддаются классификации.

Клинические проявления атаксии Фридрейха были описаны у 115 пациентов из 90 семей (Harding, 1981b). Основные проявления представлены в таблице ниже.
Заболевание чаще всего начинается в возрасте 5-16 лет, в редких случаях — в возрасте 2-5 лет. Прогрессирующая атаксия нижних конечностей с нарушением походки является основным проявлением, в то время как поражение верхних конечностей, приводящее к неуклюжести, в ранние сроки отмечается только в 25% случаев.
Сколиоз, тремор и изменения со стороны сердца редко являются первыми проявлениями, но формируются со временем, особенно при раннем начале заболевания. Конская стопа также является ранним симптомом. При осмотре в 70-95% случаев выявляется отсутствие глубоких рефлексов.
Дизартрия, пирамидные знаки со стороны ног и утрата глубокой и вибрационной чувствительности могут появляться позже, но относятся к постоянным симптомам. Нистагм встречается нечасто (20% случаев), медленные изломанные следящие движения глаз выявляются в 12% случаев (Harding, 1981b). Нестабильность фиксации является типичным проявлением (Alper и Narayanan, 2003).
Нередко встречается атрофия зрительного нерва, а глухота отмечается только у 10% пациентов. Дистальная атрофия отмечается практически в половине случаев. Интеллект не страдает.
Поражение сердца по результатам ЭКГ обнаруживается в трети случаев и даже чаще, если ЭКГ проводится систематически. Изменения зубца Т и аномалии сегмента ST являются ранними признаками сердечной недостаточности и единственным ее проявлением.
В конечном счете формируется прогрессирующая сердечная недостаточность или аритмия с фибрилляцией предсердий, половина пациентов умирает от сердечной недостаточности (Leone et al., 1988).
Течение заболевания медленное, но прогрессирующее. В среднем пациенты утрачивали способность ходить к 25 годам, со средней продолжительностью заболевания 15,5 лет.
Сахарный диабет является дальнейшим осложнением и развивается у 10% пациентов. Он имеет тенденцию сочетаться с атрофией зрительного нерва, в некоторых случаях диабетическая кома является причиной смерти.
Атипичные формы включают легкие случаи, которые, возможно, связаны с одним и тем же локусом 9-й хромосомы. К данной группе относятся случаи сохранения сухожильных рефлексов (Palau et al., 1995) и поздние формы с началом в раннем взрослом возрасте (De Michele et al., 1994).
Результаты одной из недавних работ, в которой использовалось возможное выявление мутантного гена, предполагают, что клиническая картина более вариабельна, чем считалось раньше (Palau et al., 1995; Pandolfo 2003). 25% пациентов в рамкам одного крупного исследования имели одно или более атипичное проявление (начало после 25 лет, сохранение или даже оживление сухожильных рефлексов или отсутствие симптома Бабинского).
Возраст начала превышал 20 лет у 19 из 114 пациентов (De Michele et al., 1994). Сохраненные рефлексы среди пациентов, которые в остальном соответствуют всем критериям, зарегистрированы у значимого числа пациентов (Palau et al., 1995). Большая часть случаев, ранее отнесенных к рано начинающейся атаксии с сохранением глубоких сухожильных рефлексов (состояния, отличного от атаксии Фридрейха) (Harding, 1981b; Klockgether et al., 1991), по результатам молекулярно-генетических исследований, вероятно, имеют отношение к болезни Фридрейха.
Эта группа, очевидно, была гетерогенной как при раннем ( 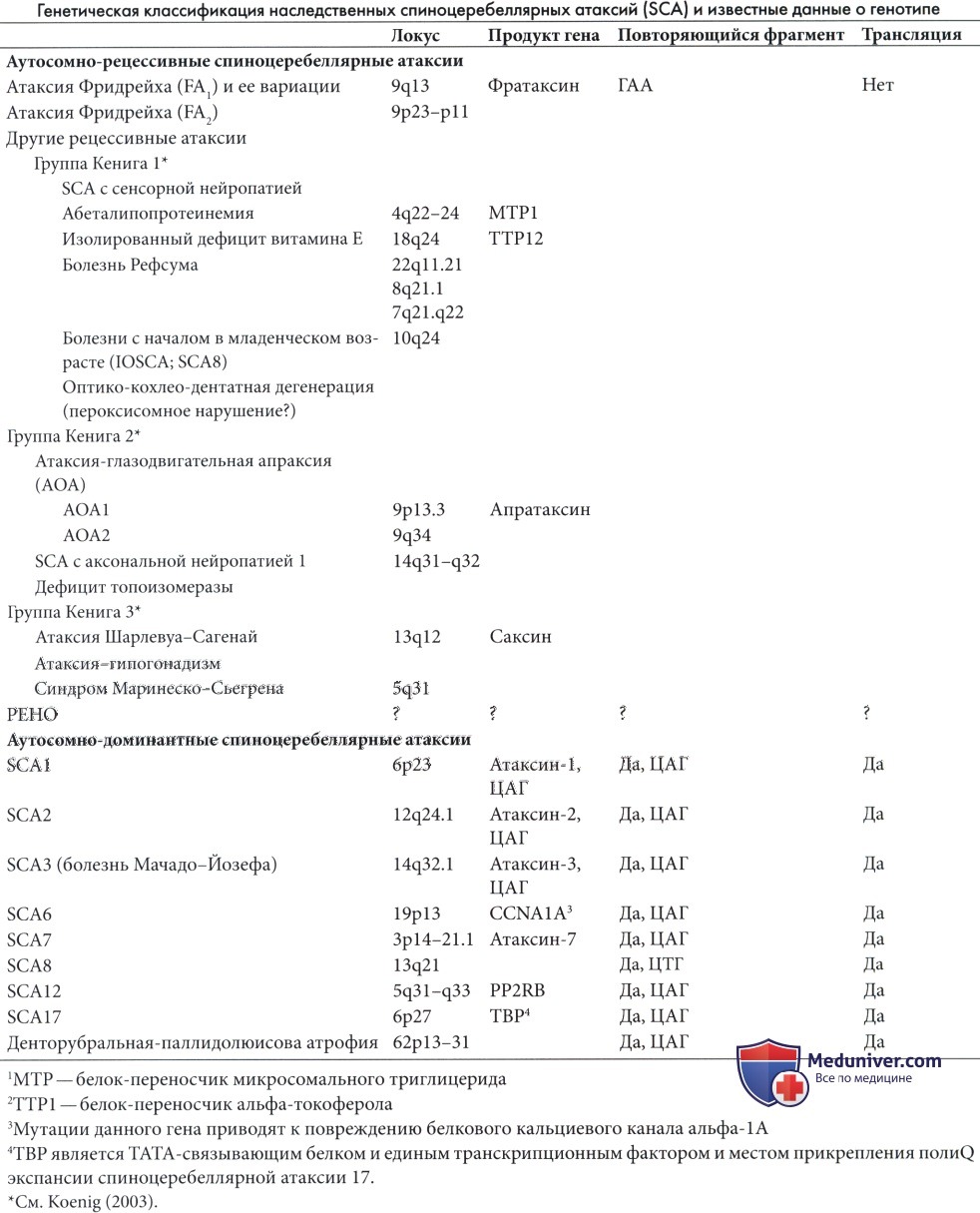
Вторая подгруппа Кенига включает атаксию со зрительной моторной апраксией (АОА), которая делится на два типа: AOA1 является одной из наиболее распространенных форм детского возраста и описана вместе с атаксией-телеангиэктазией, несмотря на то, что ее физиология кажется более сходной с спиноцеребеллярными атаксиями (SCA, см. далее). Одним из важных биологических признаков является гипоальбуминемия, которая практически постоянно обнаруживается и имеет диагностическую значимость. АОА 2 типа встречается реже и начинается позже (в позднем подростковом или раннем взрослом возрасте). Умеренно повышенный уровень альфа-фетопротеина отмечается в 75% случаев.
Клинические проявления AOA1 очень напоминают проявления атаксии-телеангиэктазии, но без признаков экстраневрологических поражений.
В отличии от атаксии-телеангиэктазии, AOA1 не связана с повышением уровня альфа-фетопротеина, хромосомными аномалиями, склонностью к раковым опухолям или повышенной радиочувствительностью культуры фибробластов (Le Ber et al., 2005). Редким, но интересным состоянием является спиноцеребеллярная атаксия с аксональной нейропатией 1 (SCA1), которая фактически является нарушением репарации ДНК, вызванной отсутствием фермента топоизомеразы-фосфодиэстеразы-1 (TDP1) (E1-Khamisy et al., 2005).
В третьей подгруппе Кенига четко описана атаксия Шарлевуа-Сагеней. Изначально синдром был описан в Квебеке, но с тех пор регистрировался и в других частях света (Gucuyener et al., 2001). Заболевание связано с мутацией гена сакцина на 13-й хромосоме (Engert et al., 2000). Фенотипические проявления включают заметную спастичность и постоянное наличие полос на глазном дне с преобладанием миелиновых волокон, радиально расходящихся от диска зрительного нерва.
Два редких аутосомно-рецессивных синдрома включают очень медленно прогрессирующую атаксию и могут рассматриваться вместе с SCA. Несмотря на то, что патология и механизмы заболеваний отличаются, они проявляются несколькими общими клиническими симптомами.
Синдром Маринеску-Шегрена включает атрофию мозжечка, преимущественно затрагивающую червь, раннее начало медленно прогрессирующей атаксии, катаракту, легкую задержку умственного развития, иногда гипогонадизм (Sewry et al., 1988) и позднее развитие специфической миопатии (Superneau et al., 1987). Заболевание развивается в результате мутации гена SLI1, кодирующего белок-шаперон, ключевой регулятор основных функций эндоплазматической сети.
РЕНО синдром (прогрессирующая энцефалопатия с периферическими отеками, гипсаритмией и атрофией зрительного нерва, также называемая церебелло-оптический синдром) является рецессивным заболеванием, зарегистрированным преимущественно в Финляндии (Salonen et al., 1991), хотя регистрировались случаи и в других странах. Основными проявлениями являются рано начинающиеся припадки, легкий дисморфизм, периферические отеки и регрессия, начинающаяся в возрасте 3-5 месяцев. Атрофия зрительного нерва развивается к концу первого года (Haltia и Somer, 1993).
Видео этиология, патогенез атаксии Фридрейха
— Вернуться в оглавление раздела «Неврология.»
Редактор: Искандер Милевски. Дата обновления публикации: 12.3.2021
Атаксия Фридрейха
Атаксия Фридрейха — наследственное нейродегенеративное заболевание, для которого характерно нарушение выведения ионов железа из околомитохондриального пространства клетки.
Среди европейцев распространенность заболевания составляет 1:20 000–1:50 000, а во всем мире каждый 120-й житель имеет предрасположенность к данной патологии. Причиной развития атаксии Фридрейха является мутация в гене FXN, в частности, нестабильное увеличение триплетов GAA. Данный ген кодирует специфический белок фратаксин, который отвечает за транспорт ионов железа из околомитохондриального пространства и не допускает тем самым образования свободных радикалов, которые оказывают выраженное повреждающее действие на центральную и периферическую нервную систему, а также другие органы.
Наследование атаксии Фридрейха проходит по аутосомно-рецессивному типу. Возможно бессимптомное носительство гена.
Клинические проявления
Мутации в гене FXN не сразу приводят к выраженному клиническому проявлению атаксии Фридрейха. Заболевание может не давать о себе знать десятилетиями, но обычно первые признаки возникают в в первом десятилетии. Дебют болезни начинается с расстройств походки и координации движений. Пациент предъявляет жалобы на неуверенность, шаткость, неловкость при движениях, отмечает частые падения. Позже присоединяются расстройства движений верхних конечностей, появление тремора. Среди других проявлений атаксии Фридрейха отмечаются:
Кроме того, заболевание сопровождается различными нарушениями работы сердца, например, аритмией, в тяжелых случаях — сердечной недостаточностью. Часто у пациентов с атаксией Фридрейха отмечаются деформации костей.
Диагностика атаксии Фридрейха
Поставить точный диагноз в некоторых случаях бывает затруднительно. Пациент может длительное время наблюдаться у невролога, кардиолога, ортопеда или других специалистов, которые не всегда могут заподозрить атаксию Фридрейха. Для того чтобы выявить характерные изменения, требуется пройти комплексное обследование, в план которого будут входить следующие методы:
Большое значение в диагностике атаксии Фридрейха имеет генетическое обследование, с помощью которого можно вывить мутацию в гене FXN и достоверно подтвердить наличие заболевания. Пройти такое обследование можно в медико-генетическом центре «Геномед».
Эффективного лечения, которое смогло бы устранить причину атаксии Фридрейха, в настоящее время не разработано. Однако для улучшения качества и продолжительности жизни может применяться симптоматическая терапия, которая всегда подбирается индивидуально. Для нормализации работы митохондрий назначаются антиоксиданты, стимуляторы активности дыхательной цепи, кофакторы энзимных реакций. Деформации костей исправляются преимущественно хирургическими методами. Для коррекции эндокринных нарушений применяются гормоны.
Для того чтобы замедлить прогрессирование атаксии Фридрейха, может назначаться ЛФК, при необходимости подбираются протезы и инвалидные кресла, которые помогут пациенту сохранить активный образ жизни.
Атаксия Фридрейха является неизлечимым прогрессирующим заболеванием. Прогноз для жизни пациента во многом зависит от возраста, в котором оно развилось, и симптомов. У женщин течение более благоприятное, чем у мужчин. Наиболее опасными считаются осложнения в виде сахарного диабета, сердечной недостаточности, бронхопневмонии. При отсутствии данных нарушений пациенты могут дожить до 70 лет и более, в противном случае продолжительность жизни ограничивается 20 годами с момента начала прогрессирования болезни.
Семейная атаксия Фридрейха, болезнь Фридрейха
1. Общие сведения
Атаксия (древнегреч. «неупорядоченность») – неврологический патологический феномен, заключающийся в рассогласованности, хаотичности моторики: нарушаются сложные нейромышечные взаимосвязи, в результате чего движения больного становятся нецеленаправленными и непродуктивными. Атаксия, выраженная в той или иной степени, сопутствует многим заболеваниям центральной нервной системы (ЦНС), – например, атрофическим и опухолевым, а также некоторым психопатологическим расстройствам, – однако выделяется группа атаксий как самостоятельных болезней.
В частности, известно более двух десятков вариантов наследственной спиноцеребеллярной (т.е. обусловленной сочетанным поражением спинного мозга и мозжечка) атаксии, или SCA, представляющей собой генетически обусловленный дегенеративно-дистрофический процесс в указанных структурах ЦНС.
Семейная атаксия Фридрейха (названа по имени врача, который в 1860 году дал первое клиническое описание болезни) является наиболее распространенным из наследственных нейродегенеративных заболеваний с преобладанием атаксии в клинической картине. Встречается, по различным источникам и в некоторой региональной зависимости, у 3-7 человек на каждые сто тысяч населения; пассивное носительство дефектного гена распространено гораздо шире (около 1%). От пола вероятность запуска процесса не зависит.
2. Причины
Как указано выше и как следует из названия, семейная атаксия Фридрейха является наследственным заболеванием. К настоящему времени выявлен и идентифицирован участок генома, ответственный за начало и прогрессирование специфической симптоматики; при одной или нескольких мутациях на этом участке 9-ой хромосомы нарушается кодирование фратаксина – одного из белков, который регулирует, в частности, усвоение железа и выведение из организма избыточных его соединений. При дефиците или в отсутствие фратаксина образуются свободные радикалы, с участием которых протекают разрушительные для нейронных тканей биохимические реакции. Признаки органического поражения ЦНС обнаруживаются во многих нервных сплетениях и пучках спинного мозга, в коре и подкорковых зонах головного мозга, а также в мозжечке, – что и формирует характерную клиническую картину.
3. Симптомы
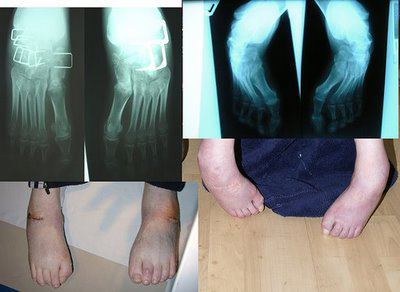
Семейная атаксия Фридрейха, как правило, манифестирует в дошкольном или допубертатном периоде, однако описаны случаи значительно более позднего дебюта, – в молодом или зрелом возрасте. Первыми симптомами обычно становятся неуверенность, «неуклюжесть», шаткость походки; затем нарушается мелкая моторика рук, ухудшается почерк, речь становится невнятной в связи с постепенно прогрессирующими нарушениями артикуляции. Больные отмечают нарастающую слабость в мышцах, ослабление чувствительности (в частности, к вибрации). На поздних стадиях мышечные ткани атрофируются, больные утрачивают способность к самостоятельному передвижению. Нейродегенеративные процессы приводят также к атрофии зрительного нерва и прогрессирующему снижению зрения, сфинктерным нарушениям, деменции (распаду интеллектуально-мнестических функций). Типичным и в конечном счете фатальным почти всегда оказывается присоединение сердечнососудистой патологии, тяжелых инфекций, сахарного диабета. Характерна также деформация скелетных структур (т.н. стопа Фридрейха); при начале в раннем детском возрасте семейная атаксия этого типа приводит к глубоким задержкам полового созревания.
Нарушения, а затем и выпадение ряда рефлексов, аномальное развитие опорно-двигательного аппарата, кардиологические и эндокринные расстройства зачастую указывают на семейную атаксию Фридрейха задолго до того, как появится собственно атактическая симптоматика. Диагноз уточняется и подтверждается с учетом результатов медико-генетического исследования, электронейромиографической диагностики, рентгенографии, ЭКГ, ЭхоКГ и других инструментальных методов.
4. Лечение
Этиопатогенетической терапии или средств профилактики на сегодняшний день нет; все принимаемые меры (даже радикальные, например, ортопедическое хирургическое вмешательство) носят лишь паллиативный характер и направлены на смягчение доминирующей симптоматики. Прогноз, в целом, неблагоприятный: неуклонное прогрессирование нейродегенеративного процесса результирует летальным исходом через 15-20 лет от манифестных проявлений. Однако в отдельных случаях, когда пациент находится под постоянным терапевтическим контролем и удается затормозить развитие тяжелой сердечной, легочной, эндокринной недостаточности, больные доживают до старости.
Атаксия Фридрейха
Атаксия Фридрейха — это наследственное заболевание аутосомно-рецессивного типа (активизируется, если ребенок унаследовал ген заболевания от обоих родителей), при котором происходит мутация гена, отвечающего за кодировку белка — фратаксин (белок митохондрий, который отвечает за вывод железа) и гибель нейронов, β-клеток островков Лангерганца поджелудочной железы, сетчатки, костно-мышечной системы, кардиомиоцитов.
Факт поражения проводящих путей спинного мозга в нервной системе до конца не изучен и причины данной патологии малоисследованы. Свое название заболевание получило в честь немецкого врача, который занимался исследованием его природы. В целом, атаксия характеризуется несогласованностью движения мышц.
Заболевание встречается довольно редко (2-5 случаев на 100 тысяч), что усложняет как его диагностику, так и само лечение. При этом статистика называет одного носителя на 120 человек. Считается, что этот вид патологии наиболее часто встречаемый среди наследственных атаксий. Негроидная раса не подвержена данной патологии по неизвестным причинам.
Причины атаксии Фридрейха
Данную патологию человек может получить только в случае, если оба его родителя будут носителями мутирующего гена. Мутация происходит в длинном плече девятой хромосомы, что и провоцирует нарушения в синтезе белка фратаксина из митохондрий, которые, в свою очередь, выполняют роль «клеточных энергетических станций».
В митохондриях накапливается железо и окисляется. Происходит транспорт кислорода в организме. При нарушении синтеза железа, его количество в митохондриях резко и значительно возрастает (примерно в десять раз). При этом клеточное железо остается в пределах нормы, а уровень цитозольного железа падает.
Такие процессы приводят в активацию гены, которые кодируют фрагменты, отвечающие за транспортировку железа — ферроксидаз и пермеаз. Баланс внутриклеточного железа нарушается еще больше. В результате высокой концентрации железа в клетке активизируются радикалы, которые обладают повреждающим действием и разрушают клетку изнутри. Самыми уязвимыми клетками оказываются нейроны (особенно в заднем и боковом столбах спинного мозга, в спиномозжечковых трактах, волокна периферических нервов).
Учитывая степень мутации гена, выделяются «классические» формы заболевания и атипичные, так сказать облегченные версии, доброкачественные синдромы.
Наследственная атаксия Фридрейха — самая распространенная среди атаксий.
Симптомы атаксии Фридрейха
Клиническая картина более четко разворачивается в возрасте от 10 до 20 лет, хотя не исключено выявление симптоматики атаксии Фридрейха и в более позднем возрасте. Есть гипотеза, что классическая и атипичная формы данного заболевания могут быть вызваны различными мутациями одного или нескольких генов. Первые симптомы чаще всего проявляются в период становления репродуктивной системы.
Клиническая картина характеризуется сочетанием неврологических и экстраневральных симптомов. До появления ДНК диагностики клиническая картина заболевания описывалась только в классической форме. Позднее ученые пришли к выводу, что спектр заболевания намного глубже, а распространённость выше, поэтому стали выделять стертые и атипичные формы атаксии Фридрейха.
Среди неврологических симптомов при атаксии Фридрейха выделяют:
К экстраневральным симптомам относятся:
Нередко электрокартографическая симптоматика значительно опережает неврологические признаки атаксии Фридрейха (иногда на несколько лет), поэтому бывает очень сложно правильно диагностировать данное заболевание. Пациенты чаще всего длительное время стоят на учете у кардиолога с диагнозом ревмокардит.
Важными для диагностики признаками данного заболевание являются также скелетные деформации:
Такие признаки, как и кардиомиопатия, могут появиться задолго до неврологических симптомов.
При атаксии Фридрейха наблюдается расстройство эндокринной системы, которое может проявляться в виде следующих заболеваний:
Очень часто у пациентов с атаксией Фридрейха обнаруживается катаракта, поэтому ее также считают частью клинической картины данного заболевания.
Атаксия Фридрейха характеризуется быстрым прогрессированием и наращиванием симптоматики. Длительность заболевания зачастую составляет не более двадцати лет.
Выраженная клиническая картина атипичной атаксии Фридрейха наблюдается позже, чем в классической форме — примерно на третьем-пятом десятке жизни человека.
Течение происходит в более облегченной форме, чем при классической атаксии и исход заболевания более благоприятен:
Такие клинические случаи описываются под названием «поздняя атаксия Фридрейха» или «атаксия Фридрейха с сохраненными рефлексами».
Диагностика атаксии Фридрейха
Диагностика этого генетического заболевания сложная. Экстраневральная симптоматика без неврологических признаков зачастую усложняет диагностирование. Сопутствующие заболевания (сахарный диабет, кардиомиопатия и др.) лечат как индивидуальные болезни, а не признаки атаксии Фридрейха.
Компьютерная томография
Отсутствие адекватного лечения ускоряет процесс прогрессирования заболевания и приводит его в тяжелую стадию. Основным диагностическим методом всех атаксий считается компьютерная томография головного мозга. Но в данном случае она малоэффективна, так как большинство изменений в мозге при атаксии Фридрейха обнаруживаются только на поздних стадиях. Это объясняется спинальной локализацией изменений. Ранние стадии заболевания не видны при КТ. Зачатую в поздних стадиях можно обнаружить только незначительные атрофии мозжечка и полушарий, некоторое расширение мозговых цистерн, боковых желудочков, субарахноидального пространства.
МР-томография
Назначается МР-томография, с помощью которой удается обнаружить атрофию в спинном мозге на ранних стадиях, а также исследуются поперечные размеры спинного мозга. При атаксии Фридрейха они ниже нормы. Видна также умеренно выраженная атрофия моста, мозжечка и продолговатого мозга.
С помощью электрофизиологического исследования устанавливается степень поражения чувствительности нервов конечностей. При атаксии Фридрейха значительно снижена или вовсе отсутствует амплитуда потенциалов действия чувствительности нервов конечностей.
Лабораторные исследования
Назначаются также лабораторные тесты — анализ крови на выявление толерантности к глюкозе. Этот анализ проводится как возможность исключения или подтверждения одного из сопутствующих заболеваний — сахарный диабет. Проводится лабораторный тест на исследование гормонов. Назначается рентгеноскопическое исследование позвоночника.
ДНК исследование
Важнейшим методом диагностирования атаксии Фридрейха является ДНК-диагностика. Для этого сравниваются образцы крови пациента с образцами обоих родителей и ближайших кровных родственников. Это заболевание может быть выявлено у плода уже на 8-12 неделе внутриутробного развития. Проводится также ДНК-диагностика ворсинок хориона. Иногда для выявления данного заболевания у плода берется амниотическая жидкость (на 16-24 неделе).
Одним из необходимых методов диагностики заболевания Фридрейха считается ЭКГ. Выявленные аритмия сердца, патологии межжелудочковой перегородки подтверждают диагноз. Сложность диагностирования заключается в том, что симптомы поражения сердечно-сосудистой системы могут появиться значительно раньше неврологических (иногда на несколько лет). Часто пациенты становятся на учет у кардиолога с диагнозом «ревмокардит».
Дифференциальная диагностика
Для объективности диагноза пациент обязательно проходит консультацию нескольких врачей: эндокринолога, офтальмолога, ортопеда, кардиолога.
Диагностика этого генетического заболевания — непростой процесс из-за сложности дифференцировать заболевание в ряде других, практически идентичных, а часто и сопутствующих заболеваний:
Лечение атаксии Фридрейха
Так как заболевание наследственное, то весь процесс лечения сводится к задержке прогрессирования болезни. Это в большинстве случаев позволяет пациенту длительное время вести активный способ жизни и избегать осложнений.
Для лечения атаксии Фридрейха назначают прием метаболических лекарственных препаратов, которые бывают трех видов:
Могут назначаться также препараты, которые питают сердечную мышцу и улучшают ее метаболизм.
В некоторых случаях необходим прием бутолотоксина — препарата, устраняющего мышечные спазмы.
Важным звеном в лечении считается ЛФК. Особое внимание уделяется тренировкам мышц и координации движений. Правильно подобранный комплекс упражнений помогает избавиться от болезненных ощущений при движении.
Иногда приписывается диетическое питание. Принцип диеты заключается в ограничении употребления углеводов, переизбыток которых провоцирует симптоматику.
Прогноз атаксии Фридрейха
Заболевание неизбежно имеет прогрессирующее течение, заканчивающееся смертью пациента в результате дыхательной или сердечной недостаточности.
Половина пациентов не доживает до сорока лет. Диагностируются случаи, когда больному удавалось пережить семидесятилетие. Это может произойти при условии отсутствия заболеваний сердца и сахарного диабета.