Наследственная аутосомно рецессивная несиндромальная нейросенсорная тугоухость что такое
Молекулярно-генетическая диагностика несиндромальной нейросенсорной тугоухости, связанной с мутациями в гене коннексина 26. Используется для подтверждения диагноза или для определения статуса носителя. Проводится полное исследование последовательности гена GJB2, что позволяет обнаружить все возможные мутации, приводящие к развитию заболевания.
Изолированная тугоухость или глухота, врождённая нейросенсорная (сенсоневральная) тугоухость, аутосомно-рецессивная несиндромальная нейросенсорная тугоухость, недифференцированная глухота.
DFNB1A(Deafness, autosomal recessive 1A).
Какой биоматериал можно использовать для исследования?
Буккальный (щечный) эпителий, венозную кровь.
Как правильно подготовиться к исследованию?
Подготовки не требуется.
Общая информация об исследовании
Более 80 % всех случаев наследственных форм нарушения слуха связано с несиндромальной нейросенсорной тугоухостью.
Тугоухость – это снижение слуха, затрудняющее речевое общение. Нейросенсорная тугоухость обусловлена повреждением чувствительных нервных клеток внутреннего уха, слухового нерва и центральных образований слуховой системы.
При несиндромальной форме снижение слуха не сопровождается заболеваниями других органов и систем, которые наследуются вместе с тугоухостью.
Частой причиной наследственных несиндромальной тугоухости и глухоты является мутационное повреждение генов белков-коннексинов.
Одним из самых значимых в развитии тугоухости является ген GJB2, который кодирует белок коннексин 26 (Cx26). Этот трансмембранный белок участвует в образовании межклеточных контактов – коннексонов – в тканях внутреннего уха.
Почти все клетки улитки (части внутреннего уха) соединены между собой посредством щелевых контактов, состоящих из каналов для прямого межклеточного обмена ионами и молекулами, в основном для циркуляции К+(ионов калия) в улитке. Межклеточные ионные каналы обеспечивают таким образом передачу сигналов между клетками, участвующими в восприятии звука.
Основой патогенеза нарушений слуха при мутациях в Сх26 является первичное отсутствие в тканях улитки специфического белка, необходимого для построения межклеточных каналов, формирующих щелевые контакты. Поэтому функция органа слуха на клеточном уровне, в зависимости от типа мутации, становится нарушенной в разной степени либо совсем невозможной. В 80 % случаев отмечается тугоухость четвертой степени, в 17 % – третьей степени и только в 3 % – второй степени.
Нейросенсорная несиндромальная тугоухость выявляется у ребенка в доречевой период, реже в период от рождения до 6 лет и позднее 6 лет.
Для тугоухости, связанной с нарушениями в коннексине 26, характерно стабильное течение. Но в течение жизни качественные изменения фенотипа (изменение степени тяжести) под влиянием внешних воздействий могут накладываться на те, что связаны с мутациями в гене. Возможно ухудшение слуха у ребенка после простудных заболеваний, вирусных инфекций, прививок, черепно-мозговых травм.
Тяжелая потеря слуха способна приводить к задержке речевого развития разной степени выраженности, нарушению психоэмоционального и социального развития ребенка и является основанием для получения инвалидности.
Обнаружение мутаций гена GJB2 свидетельствует о том, что тугоухость имеет наследственный характер (так как мутации наследуются от родителей) и в дальнейшем будет передаваться потомству.
Рождение ребенка с нарушением слуха можно предупредить, проверив себя на носительство мутаций в гене коннексина 26. При планировании детей необходимо знать о наличии возможной патологии в гене коннексина 26 у себя и у партнера, потому что если мутация одна, то она никак не проявляется (носитель здоров). Но заболевание наследуется по аутосомно-рецессивному типу, и в случае когда в генотипе плода встретятся две мутации гена GJB2, унаследованные от родителей (здоровых носителей), вероятность рождения ребенка с нарушением слуха будет составлять 25 %.
Если в семье среди родственников есть (или были) случаи изолированного нарушения слуха с рождения (или с детства) необходимо пройти генетическое исследование на носительство мутации гена коннесксина 26.
При обнаружении мутации в гене коннексина 26 рекомендуется консультация у генетика.
Генетический скрининг новорождённых на мутации в гене GJB2 (по данным зарубежных авторов) способствует раннему выявлению наследственных нарушений слуха и, следовательно, раннему излечению и реабилитации.
На сегодняшний день оптимальным методом лечения таких нарушений является кохлеарная имплантация, а после операции важна работа сурдопедагога с ребенком.
Когда назначается исследование?
Что означают результаты?
В ходе анализа проводится полное исследование последовательности гена, что позволяет обнаружить все возможные мутации, приводящие к развитию заболевания.
Существуют инактивирующие мутации, которые приводят к преждевременному прекращению синтеза белка и имеют значительные, тяжелые последствия на тканевом уровне, приводящие к дегенерации и полностью нарушающие функцию органа. Наличие в генотипе двух инактивирующих мутаций соответствует самым тяжелым нарушениям слуха. К таким мутациям относится мутация 35delG, отвечающая за 51 % всех случаев врождённой и ранней нейросенсорной тугоухости. Так, для гомозигот по мутации 35delG гена GJB2 характерна врождённая тяжелая тугоухость с началом проявлений от момента рождения до первого, реже второго года жизни.
Благодаря проведенным исследованиям известно, что в ряде регионов нашей страны каждые 2 человека из 100 являются носителями мутации 35delG, поэтому вероятность рождения ребенка от носителей измененного гена очень высока.
Второй вид мутаций – неинактивирующие, приводящие к замене одной аминокислоты в последовательности белка, что отражается на его функции. При таких мутациях структура коннексиновых каналов может быть сформирована правильно, но они оказываются функционально неполноценными (или изменяется их проницаемость).
Неинактивирующие мутации приводят к меньшей потере слуха.
Как правило, дети успевают приобрести речевые навыки даже без помощи слухового аппарата. Это означает, что поиск мутаций в гене GJB2 следует проводить также при менее тяжелых формах тугоухости и при поздней манифестации заболевания.
Генетические маркеры
Нейросенсорная несиндромальная тугоухость
OMIM 220290
Наша команда профессионалов ответит на ваши вопросы
Что такое наследственная тугоухость?
Какой тип наследственной тугоухости наиболее часто встречается?
Около 75% всех случаев наследственной тугоухости относятся к рецессивным несиндромальным нарушениям слуха (РННС) или рецессивной несиндромальной тугоухости.
При рецессивном типе наследования ребенок получает от каждого из родителей один и тот же вариант гена, который вызывает данную форму нарушения слуха. «Рецессивный» ген проявляется лишь в паре с другим таким же геном и вызывает РННС. При этом родители ребенка не страдают нарушением слуха, так как они имеют нормальный вариант данного гена в паре генов, полученных от своих родителей. Тем не менее, они являются носителями гена рецессивной несиндромальной глухоты. Таким образом, у ребенка может быть нарушение слуха, тогда как его родители и все другие родственники могут иметь нормальный слух в любом возрасте.
Под несиндромальной формой понимают то, что снижение слуха не сопровождается другими признаками или заболеваниями других органов и систем, которые передавались бы по наследству вместе с тугоухостью, что имеет место при синдромальных формах. Например, синдром Пендреда – самый частый синдромальный вариант тугоухости – рецессивное заболевание, характеризующееся сочетанием нарушения слуха с формированием эутиреоидного зоба. Зоб формируется в подростковом возрасте и позже, поэтому у детей дифференциальная диагностика синдрома Пендреда и несиндромальной рецессивной тугоухости крайне трудна.
Аутосомно-доминантные формы несиндромальной тугоухости встречаются относительно редко, и составляют не более 25% всей несиндромальной глухоты. При доминантных формах достаточно одной измененной копии гена для того, чтобы заболевание проявилось. В таких случаях, как правило, у ребенка с тугоухостью болен один из родителей, однако возможно и возникновение новой мутации.
Каким образом ребенок получает РННС?
Время от времени воздействие каких-либо факторов может вызывать изменение гена. Генетики называют это изменение мутацией. Многие люди и не подозревают о том, что они являются носителями измененных генов. Данные изменения, однажды возникнув, передаются по наследству из поколения в поколение. Большинство мутаций не влияют на состояние организма, но иногда некоторые из них в силу ряда причин проявляют свое действие. Одной из причин является встреча двух носителей одного и того же измененного гена. Они могут стать родителями ребенка с рецессивной несиндромальной глухотой. Этот ребенок получает измененный ген от каждого из родителей и, таким образом, будет иметь две копии измененного гена. Только в данном случае из-за отсутствия нормального варианта гена мутация проявляет свое действие. Для этих родителей риск рождения ребенка с врожденной несиндромальной глухотой составляет 25%. Как правило, степень потери слуха изначально достаточна для коррекции и обучения. Дети с нормальным слухом в данном браке могут родиться в 75% случаев, причем часть детей могут иметь здоровый генотип (25%) и им в будущем ничто не угрожает, а другие, как и их родители, являются носителями измененного гена (50%) и для них ситуация может повториться.
Брак двух слышащих родителей, носителей измененного гена
Самым значимым для развития тугоухости оказался ген коннексина-26 (GJB2). Только одно изменение в этом гене, которое обозначается как мутация 35delG, отвечает за 51% всех случаев врожденной и ранней детской тугоухости. Известны и другие изменения в этом гене. Благодаря проведенным исследованиям известно, что в нашей стране каждый 20 житель является носителем мутации 35delG. Поэтому, как это ни печально, вероятность встречи носителей измененного гена достаточно высока.
Какая польза ожидается от идентификации генов, приводящих к РННС?
Это возможность точного определения причин нарушения слуха. Генетический анализ позволяет сделать правильный прогноз повторения заболевания в семье, а также оценить вероятность рождения детей с нарушением слуха в семьях родственников. Раннее выявление генетического дефекта помогает своевременно выбрать правильную тактику лечения и реабилитации ребенка с нарушением слуха, позволит уберечь его от приема ненужных лекарств.
Даже здоровый человек может узнать свой генотип, так как он может оказаться носителем гена тугоухости (с вероятностью 1/20). Исследование на носительство гена тугоухости особенно актуально для лиц, имеющих родственников с нарушением слуха, а также супругов, состоящих в близкородственном браке.
Для семейной пары, в которой оба являются носителями мутации в гене тугоухости (вне зависимости от того, есть ли у них уже ребенок с нарушением слуха или нет) есть возможность проведения пренатальной (дородовой) ДНК-диагностики заболевания у плода на раннем сроке беременности (9-12 недель).
Каким образом проводится поиск генов, приводящих к РННС?
У пациентов с ННС без мутаций в гене GJB2 наблюдаются мутации в других генах: описано еще более 100 генов, ответственных за ННС. В Центре Молекулярной Генетики проводится поиск мутаций 32 наиболее частых генетических форм ННС и маскирующихся под них синдромов у пациентов без мутаций в гене GJB2 методом секвенирования панели 32 генов, представленных в Таблице.
Заболевание
OMIM
STRC
DFNB16, с.глухоты и мужского бесплодия
Наследственная аутосомно рецессивная несиндромальная нейросенсорная тугоухость что такое
Что такое наследственная тугоухость?
Наследственная тугоухость – нарушение слуха, наследуемое в семье. Причиной является наследование измененных генов. При этом семья может знать, а может и не знать, о случаях тугоухости в других поколениях. Вероятно, Вы уже знаете, что гены являются носителями наследственной информации, которая определяет развитие целого организма из одной оплодотворенной яйцеклетки. Каждый ген в отдельности отвечает за образование определенного белка и может иметь несколько вариантов своего строения. Одни варианты являются нормальными, а другие приводят к развитию патологии. Все мы получаем по две копии каждого гена – одну от матери, другую от отца. Таким образом, каждый человек имеет два варианта одного и того же гена. Ряд генов в организме отвечает за образование и работу органа слуха. В общей сложности таких генов не менее 100. Не удивительно, что согласно данным последних исследований более 50% всех случаев врожденной и ранней детской тугоухости связано с наследственными причинами. Считается, что каждый восьмой житель Земли является носителем одного из генов, вызывающих рецессивную тугоухость.
Какой тип наследственной тугоухости наиболее часто встречается?
Около 75% всех случаев наследственной тугоухости относятся к рецессивным несиндромальным формам нарушения слуха (РННС) или рецессивной несиндромальной тугоухости.
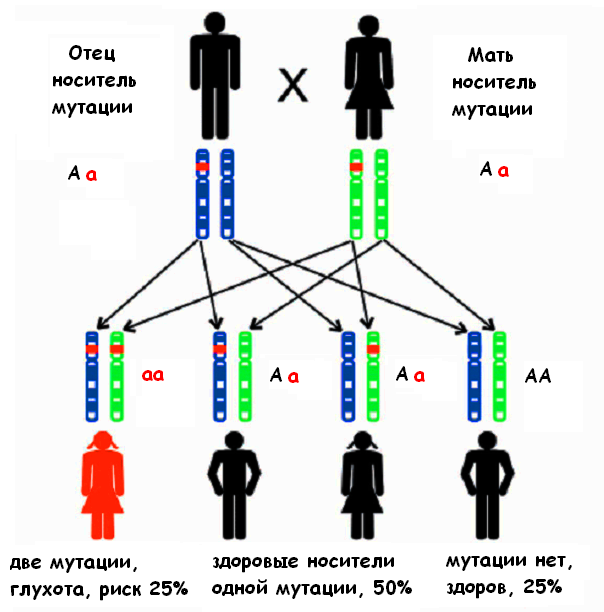
При рецессивном типе наследования ребенок получает от каждого из родителей один и тот же патологический вариант гена, который вызывает данную форму нарушения слуха (см. рис.). «Рецессивный» ген проявляется лишь в паре с другим таким же геном. При этом родители ребенка не страдают нарушением слуха, так как они имеют один нормальный вариант данного гена в паре генов, полученных от своих родителей.
Тем не менее, они являются носителями гена рецессивной несиндромальной глухоты. Таким образом, у ребенка может быть нарушение слуха, тогда как его родители и все другие родственники имеют нормальный слух в любом возрасте.
Под несиндромальной формой понимают то, что снижение слуха не сопровождается другими признаками или заболеваниями других органов и систем, которые передавались бы по наследству вместе с тугоухостью, что имеет место при синдромальных формах (например, синдром Пендреда – это синдром, характеризующийся сочетанием нарушения слуха и нарушения функции щитовидной железы).
Каким образом ребенок получает РННС?
Итак, каждый из нас получает половину наших генов от отца и другую половину от матери. Какой ген из родительской пары генов мы получаем – явление чисто случайное.
Время от времени воздействие каких-либо факторов может вызывать изменение гена. Генетики называют это изменение мутацией. Многие люди и не подозревают о том, что они являются носителями измененных генов. Данные изменения, однажды возникнув, передаются по наследству из поколения в поколение. Большинство мутаций не влияют на состояние организма, но иногда некоторые из них в силу ряда причин проявляют свое действие. Одной из причин является встреча двух носителей одного и того же измененного гена. Они могут стать родителями ребенка с рецессивной несиндромальной глухотой (см. рис.). Этот ребенок получает измененный ген от каждого из родителей и таким образом, будет иметь две копии измененного гена. Только в данном случае из-за отсутствия нормального варианта гена мутация проявляет свое действие. Для этих родителей риск рождения ребенка с врожденной несиндромальной глухотой составляет 25%.
Как правило, степень потери слуха изначально достаточна для коррекции и обучения
Дети с нормальным слухом в данном браке могут родиться в 75% случаев, причем часть детей могут иметь здоровый генотип (25%) и им в будущем ничто не угрожает, а другие, как и их родители, являются носителями измененного гена (50%) и для них ситуация может повториться.
Каким образом ребенок получает РННС?
Самым значимым для развития тугоухости оказался ген коннексина 26. Только одно изменение в этом гене – мутация 35delG, отвечает за 40% всех случаев врожденной и доречевой детской тугоухости. Известны и другие изменения в этом гене. Благодаря проведенным исследованиям показано, что в ряде регионов нашей страны каждые 4 человека из 100 являются носителями мутации 35delG. Поэтому вероятность встречи носителей измененного гена очень высока. Предварительный анализ гена при планировании потомства может выявить носительство.
Выявление носительства одной патологической мутации у здоровых лиц является поводом для беспокойства о риске врожденной тугоухости для их детей. На практике мы часто сталкиваемся с ситуацией, когда сведения о случаях в семье или об измененном генотипе тщательно скрываются из-за страха быть “виновными”, если заболевание повторится.
Исследования показали, что в 80% супружеских пар, где оба супруга страдают нарушением слуха, рождаются нормально слышащие дети, и только 20% семей имеют глухих/тугоухих детей. Это связано со значительным разнообразием причин врожденной тугоухости, в том числе и генетических. Поэтому, у тех кто имеет нарушение слуха, но хочет родить ребенка с нормальным слухом есть выбор. В современных условиях определенную помощь в этом оказывает генетический анализ. В случае, если речь идет о нарушении слуха связанном с мутациями в гене коннексина 26, зная свои генотипы будущие родители могут точно знать, что будет ли у их детей нарушение слуха.
Важно помнить, что в случае рецессивной тугоухости передается не заболевание, передается измененный ген. Заболевание проявляется только в случае встречи двух измtненных генов в генотипе человека. Если ребенку поставили диагноз наследственной тугоухости, это не значит, что всем его потомкам угрожает нарушение слуха.
Если два человека с рецессивным несиндромальным нарушением слуха (генотип аа ), которое вызвано одним и тем же геном вступят в брак, все их дети будут страдать нарушением слуха, то есть риск в данном случае составит 100%. Внешне это будет выглядеть как повторение заболевания в семье.
Если человек с рецессивной формой несиндромальной тугоухости (генотип аа ) вступит в брак с лицом, не являющимся носителем подобного гена (генотип АА), то нарушения слуха у детей этой пары не будет. Что же получается? Заболевание у одного из супругов наследственное, а повторения не произошло? Да, таковы законы генетики. В то же время, все, рожденные в такой паре дети, будут носителями измененного гена (генотип А а ). Эта ситуация характерна для слышащих детей в паре глухих родителей. Эти дети будут здоровы и не слудует опасаться развития у них нарушения слуха, по крайней мере, в следствие данной генетической причины.
В современных условиях в подобную ситуацию могут попасть дети с рецессивной несиндромальной формой нарушения слуха (генотип аа ), прошедшие кохлеарную имплантацию, которые будут выбирать партнера в слышащей среде. То есть они могут рассчитывать на то, что в большинстве случаев среди здоровых людей они выберут партнера без изменений в гене коннексина 26. Но если они встретят здорового носителя такого же гена (генотип А а ), то в этом случае у них будет высокий риск рождения глухого ребенка – 50%. Только в случае такой встречи и попадания двух измененных генов в генотип ребенка заболевание повторится.
Человек, получивший только один ген, вызывающий рецессивное несиндромальное нарушение слуха (другой ген в паре нормальный – генотип А а ), не будет иметь нарушение слуха, так как рецессивный ген при наличии нормального гена в паре не проявляет своего действия. Но такой человек будет являться носителем и имеет риск рождения глухого ребенка при встрече с таким же носителем.
Если носитель мутации гена рецессивной тугоухости (генотип А а ) вступит в брак с лицом, не являющимся носителем патологически измененного гена (генотип АА), у них не будет детей с нарушением слуха, но каждый второй ребенок может оказаться носителем измененного варианта гена. Так ген будет передаваться не проявляя себя, пока не встретит себе пару.
В 80% случаев рецесивные мутации в гене коннексина 26 приводят к тяжелой врожденной сенсоневральной тугоухости или глухоте, которые эффективно реабилитируются с помощью метода кохлеарной имплантации. В 15% случаев встречается тугоухость III степени, в 5% случаев определяется тугоухость II степени. Последние требуют слухопротезирования, при этом дети учатся в массовой школе. При легких и средних степенях данной формы тугоухости прогрессирование не характерно. Это важно помнить, так как универсальный аудиологический скрининг выявляет детей с изменениями в гене коннексина 26 и легкими нарушениями слуха уже в первые месяцы жизни, что заставляет родителей беспокоиться об ухудшении слуха в дальнейшем. Практика показывает, что такие опасения в 90% случаев не оправданы.
Скрытое носительство мутаций несиндромальной нейросенсорной тугоухости
Молекулярно-генетическая диагностика несиндромальной нейросенсорной тугоухости, связанной с мутациями в гене коннексина 26. Используется для подтверждения диагноза или для определения статуса носителя. Проводится полное исследование последовательности гена GJB2, что позволяет обнаружить все возможные мутации, приводящие к развитию заболевания.
Изолированная тугоухость или глухота, врожденная нейросенсорная (сенсоневральная) тугоухость, аутосомно-рецессивная несиндромальная нейросенсорная тугоухость, недифференцированная глухота.
DFNB1A(Deafness, autosomal recessive 1A).
Какой биоматериал можно использовать для исследования?
Буккальный (щечный) эпителий, венозную кровь.
Как правильно подготовиться к исследованию?
Подготовки не требуется.
Общая информация об исследовании
Более 80 % всех случаев наследственных форм нарушения слуха связано с несиндромальной нейросенсорной тугоухостью.
Тугоухость – это снижение слуха, затрудняющее речевое общение. Нейросенсорная тугоухость обусловлена повреждением чувствительных нервных клеток внутреннего уха, слухового нерва и центральных образований слуховой системы.
При несиндромальной форме снижение слуха не сопровождается заболеваниями других органов и систем, которые наследуются вместе с тугоухостью.
Частой причиной наследственных несиндромальной тугоухости и глухоты является мутационное повреждение генов белков-коннексинов.
Одним из самых значимых в развитии тугоухости является ген GJB2, который кодирует белок коннексин 26 (Cx26). Этот трансмембранный белок участвует в образовании межклеточных контактов – коннексонов – в тканях внутреннего уха.
Почти все клетки улитки (части внутреннего уха) соединены между собой посредством щелевых контактов, состоящих из каналов для прямого межклеточного обмена ионами и молекулами, в основном для циркуляции К (ионов калия) в улитке. Межклеточные ионные каналы обеспечивают таким образом передачу сигналов между клетками, участвующими в восприятии звука.
Основой патогенеза нарушений слуха при мутациях в Сх26 является первичное отсутствие в тканях улитки специфического белка, необходимого для построения межклеточных каналов, формирующих щелевые контакты. Поэтому функция органа слуха на клеточном уровне, в зависимости от типа мутации, становится нарушенной в разной степени либо совсем невозможной. В 80 % случаев отмечается тугоухость четвертой степени, в 17 % – третьей степени и только в 3 % – второй степени.
Нейросенсорная несиндромальная тугоухость выявляется у ребенка в доречевой период, реже в период от рождения до 6 лет и позднее 6 лет.
Для тугоухости, связанной с нарушениями в коннексине 26, характерно стабильное течение. Но в течение жизни качественные изменения фенотипа (изменение степени тяжести) под влиянием внешних воздействий могут накладываться на те, что связаны с мутациями в гене. Возможно ухудшение слуха у ребенка после простудных заболеваний, вирусных инфекций, прививок, черепно-мозговых травм.
Тяжелая потеря слуха способна приводить к задержке речевого развития разной степени выраженности, нарушению психоэмоционального и социального развития ребенка и является основанием для получения инвалидности.
Обнаружение мутаций гена GJB2 свидетельствует о том, что тугоухость имеет наследственный характер (так как мутации наследуются от родителей) и в дальнейшем будет передаваться потомству.
Рождение ребенка с нарушением слуха можно предупредить, проверив себя на носительство мутаций в гене коннексина 26. При планировании детей необходимо знать о наличии возможной патологии в гене коннексина 26 у себя и у партнера, потому что если мутация одна, то она никак не проявляется (носитель здоров). Но заболевание наследуется по аутосомно-рецессивному типу, и в случае когда в генотипе плода встретятся две мутации гена GJB2, унаследованные от родителей (здоровых носителей), вероятность рождения ребенка с нарушением слуха будет составлять 25 %.
Если в семье среди родственников есть (или были) случаи изолированного нарушения слуха с рождения (или с детства) необходимо пройти генетическое исследование на носительство мутации гена коннесксина 26.
При обнаружении мутации в гене коннексина 26 рекомендуется консультация у генетика.
Генетический скрининг новорождённых на мутации в гене GJB2 (по данным зарубежных авторов) способствует раннему выявлению наследственных нарушений слуха и, следовательно, раннему излечению и реабилитации.
На сегодняшний день оптимальным методом лечения таких нарушений является кохлеарная имплантация, а после операции важна работа сурдопедагога с ребенком.
Когда назначается исследование?
Что означают результаты?
В ходе анализа проводится полное исследование последовательности гена, что позволяет обнаружить все возможные мутации, приводящие к развитию заболевания.
Существуют инактивирующие мутации, которые приводят к преждевременному прекращению синтеза белка и имеют значительные, тяжелые последствия на тканевом уровне, приводящие к дегенерации и полностью нарушающие функцию органа. Наличие в генотипе двух инактивирующих мутаций соответствует самым тяжелым нарушениям слуха. К таким мутациям относится мутация 35delG, отвечающая за 51 % всех случаев врожденной и ранней нейросенсорной тугоухости. Так, для гомозигот по мутации 35delG гена GJB2 характерна врожденная тяжелая тугоухость с началом проявлений от момента рождения до первого, реже второго года жизни.
Благодаря проведенным исследованиям известно, что в ряде регионов нашей страны каждые 2 человека из 100 являются носителями мутации 35delG, поэтому вероятность рождения ребенка от носителей измененного гена очень высока.
Второй вид мутаций – неинактивирующие, приводящие к замене одной аминокислоты в последовательности белка, что отражается на его функции. При таких мутациях структура коннексиновых каналов может быть сформирована правильно, но они оказываются функционально неполноценными (или изменяется их проницаемость).
Неинактивирующие мутации приводят к меньшей потере слуха.
Как правило, дети успевают приобрести речевые навыки даже без помощи слухового аппарата. Это означает, что поиск мутаций в гене GJB2 следует проводить также при менее тяжелых формах тугоухости и при поздней манифестации заболевания.
Генетические маркеры
Литература
Подписка на новости
Оставьте ваш E-mail и получайте новости, а также эксклюзивные предложения от лаборатории KDLmed