Нейрофиброматозы
Нейрофиброматозы – наследственные заболевания, характеризующиеся образованием доброкачественных опухолей в коже, мягких тканях, нервной системе и внутренних органах. Выделяют 6 типов нейрофиброматозов, клинически значимы типы I и II. Общие симптомы включают нейрофибромы на коже, опухоли спинномозговых корешков, слуховых и зрительных нервов, пигментные пятна, костные деформации. Диагностика основана на данных осмотра пациентов, выявлении опухолей с помощью МРТ и КТ спинного и головного мозга, внутренних органов. Лечение симптоматическое – проводится резекция опухолей, рентгенотерапия, химиотерапия.
МКБ-10
Общие сведения
Нейрофибромы – доброкачественные опухоли, развивающиеся из оболочек нервных волокон. Чаще всего располагаются в слоях кожи и подкожной клетчатке, иногда поражают головной мозг, нервные волокна, корешки спинного мозга, мягкие ткани, внутренние органы. Нейрофиброматоз – болезнь, при которой образуются многочисленные нейрофибромы. Распространенность разных типов патологии значительно колеблется: заболеваемость 1 типом составляет 1:2 500, 2 типом – 1:50 000. Другие варианты встречаются еще реже, их точная эпидемиология не определена. Гендерной и расовой предрасположенности не выявлено. Дебют клинических проявлений возможен в любом возрасте, зависит от типа болезни.
Причины нейрофиброматозов
Образование множественных нейрофибром детерминировано генетически. При нейрофиброматозе I существует мутация гена НФ1, расположенного на длинном плече 17 хромосомы. Он относится к генам-супрессорам роста опухолевых тканей, большая часть из которых – нейроэктодермального генеза. При дефекте в гене НФ1 нарушается синтез белков, ответственных за клеточную пролиферацию. Мутации носят характер транслокаций, делеций, инверсий, точковых изменений. Больше 80% из них приводят к синтезу нефункциональных белков или к полному отсутствию белковых молекул. Наследование происходит по аутосомно-доминантному механизму с высокой степенью пенетрации: при наличии мутационного гена у одного из родителей вероятность болезни у ребенка составляет 50%, если оба родителя имеют мутацию, риск повышается до 80-90%. Известны случаи спонтанных мутаций.
Причиной нейрофиброматоза II является мутационное изменение гена НФ2, локализованного на 22 хромосоме. Он кодирует производство белка мерлина (шванномина) – супрессора опухолевого роста. Тип наследования – аутосомно-доминантный с небольшой степенью пенетрации. Передача одного мутантного гена зачастую не проявляется, поскольку второй ген обеспечивает синтез достаточного количества белков. Если он повреждается, синтез нормальных фракций мерлина прекращается, пролиферация клеток усиливается, развивается новообразование. При других типах нейрофиброматозов также существуют мутации в генах, обеспечивающих воспроизведение молекул белков-супрессоров роста опухолей.
Патогенез
Общим патогенетическим механизмом развития нейрофиброматозов является наследственно обусловленная недостаточность какого-либо белка, подавляющего процессы опухолевой пролиферации клеток в тканях нейроэктодермального происхождения. При мутации одного гена производство опухолевых супрессоров прекращается наполовину, равновесие роста и гибели клеток смещается в сторону митотического деления. Нормальный аллельный ген частично компенсирует дефицит белка. Тяжесть нейрофиброматоза определяется тем, насколько дефектный ген влияет на активность белка-супрессора – частично или полностью нарушает функциональность, полностью блокирует производство. Кроме этого, выраженность клинических признаков зависит от сохранности противоопухолевого иммунитета.
Во многих органах и тканях пациентов формируются доброкачественные опухолевые образования, состоящие из соединительной ткани и пигментных клеток. На нервных стволах образуются невриномы; на поверхности кожи – пигментированные области, жировые бляшки, расширенные сосуды; на сетчатке глаз – факоматоз. Изменяется строение костей, они остаются недоразвитыми либо чрезмерно утолщаются, искривляется позвоночный столб.
Симптомы нейрофиброматозов
Заболевания проявляются признаками поражения кожи, нервной системы. Классическим клиническим вариантом является нейрофиброматоз типа I, на долю которого приходится 90% случаев болезни. Характерный симптом – гиперпигментация. У больных при рождении или в раннем детстве появляются кожные пятна, цвет которых варьируется от светло-золотистого до коричневого «кофе с молоком». В отдельных случаях пятна имеют фиолетовый или синий оттенок. На радужке глаза обнаруживаются узелки Лиша (пятна пигмента – гамартомы) небольшого размера, белесоватые или светло-бежевые, заметные только при офтальмологическом осмотре. Являются специфическим признаком нейрофиброматоза 1, образуются по мере взросления: у детей до 4 лет распространенность составляет 22%, с 5 до 9 лет – 41%, с 10 до 19 лет – 85%, после 20 лет – 95%.
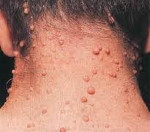
В период пубертата и позже формируются кожные и плексиформные нейрофибромы, располагающиеся соответственно подкожно (на мелких нервных волокнах, иннервирующих кожу) и на крупных нервах. Они представляют собой небольшие доброкачественные новообразования. Кожные нейрофибромы воспринимаются как косметический дефект, при определенном расположении травмируются. Плексиформные опухоли, локализующиеся по ходу периферических нервов, выявляются на конъюнктиве, веках, в брюшной полости и средостении. Проявляются хронической болью, онемением, судорогами, параличом. Опухоли ЦНС находятся внутри черепа, представлены глиомами зрительных нервных волокон, астроцитомами, эпендимомами, невриномами слухового нерва, менингиомами и нейрофибромами. Клиническая картина определяется размерами новообразований, вовлеченностью мозговых структур в патологический процесс. В детском возрасте диагностируются расстройства психического развития: снижение когнитивных способностей, гиперактивность, редко – деменция.
При тяжелом нейрофиброматозе деформируется костная система. У больных возникает сколиоз, краевые структурные изменения тел позвонков и их отростков, эрозийные поражения краев межпозвоночных отверстий и задних ребер. Характерна атрофия либо, наоборот, гипертрофия трубчатых костей. Кости часто искривлены, на поверхности обнаруживаются периостальные гребни и наслоения. В полостях костей могут образовываться нейрофибромы. Если в процесс вовлекаются кости черепа, появляется внешняя асимметрия, наиболее выраженная при поражении лицевой части и глазниц. Свод черепа может иметь атрофированные участки, дефекты, узуры, иногда отмечается локальное увеличение костного вещества.
При типе 2 формируются высокодифференцированные опухоли, которые, однако, более агрессивны, чем при заболевании 1 типа. Пигментных пятен нет. Образуются невриномы – подвижные и болезненные неоплазии. Нередко они локализуются на слуховом нерве, вызывая потерю слуха. Нейрофиброматоз 3 типа отличается большим количеством нейрофибром, ускоренным развитием нейролемм и глиом зрительного нерва, приводящих к расстройству зрения. Специфический признак – появление нейрофибром на ладонях. При болезни 4 типа симптомы похожи, сохраняется риск поражения зрительных волокон. Для 5 типа характерны пигментные темные пятна, опухоли больших размеров, провоцирующие асимметрию тела. Течение 6 типа сопровождается лишь пигментными пятнами. При 7 типе выявляются нейрофибромы средних размеров, гиперпигментации нет.
Осложнения
В 10% случаев нейрофибромы трансформируются в злокачественные опухоли. В группе высокого риска находятся пациенты с большим катамнестическим стажем, беременные женщины. У 6% детей нарушается умственное развитие: они имеют проблемы при освоении учебных навыков (чтение, письмо, счет), с трудом запоминают новую информацию, долго адаптируются в незнакомых ситуациях. Больные всех возрастов подвержены депрессии, поскольку испытывают дискомфорт, чувство стыда и неловкости из-за обезображенной внешности. Множественные нейрофибромы провоцируют эндокринные расстройства, эпилептические припадки, гипотонию мышц, стеноз почечной и легочной артерии, легочные кисты, интерстициальную пневмонию, гипертрофию клитора, нарушения развития органов ЖКТ.
Диагностика
Подозрение на нейрофиброматоз возникает при множественных подкожных опухолях, пигментных пятнах, спинальной шванноме, ухудшении слуха и зрения. Обследование проводят дерматовенеролог, невролог, офтальмолог, отоларинголог и генетик. Перед инструментальными и лабораторными процедурами осуществляется сбор семейного и личного анамнеза, клинический опрос и осмотр. В ходе генеалогического анализа выявляется передача заболевания в нескольких поколениях, реже определяется первичная спонтанная мутация. На теле пациентов обнаруживаются нейрофибромы, пигментные области (при определенных типах болезни), искривления позвоночника, деформации костей, нарушения зрения, слуха, координации движений. Производится дифференциальная диагностика различных вариантов нейрофиброматозов, исключается синдром Протея, рассеянный липоматоз, синдром Клиппеля-Треноне-Вебера. Для уточнения диагноза назначаются:
Лечение нейрофиброматозов
В настоящее время терапия данной группы заболеваний заключается в симптоматической помощи больным. Пациенты регулярно проходят обследования, нацеленные на контроль формирования и увеличения опухолей. При наличии нейрофибром, провоцирующих боль, расположенных в местах повышенного риска травмирования, сдавливающих или смещающих жизненно важные органы, проводится их хирургическое удаление. Применяются классические методики резекции неоплазий и участков нервов, криодеструкция, лазерная хирургия. При множественных новообразованиях назначается лучевая терапия, химиотерапия. Больным с поражением опорно-двигательного аппарата показаны реабилитационные мероприятия (физиолечение, ЛФК).
Активно разрабатываются способы этиологического лечения нейрофиброматозов. На стадии клинических испытаний находится терапия ингибиторами RAS (белков-активаторов роста опухолей) у лиц с нейрофиброматозом первого типа. Этап теоретических разработок проходят методы генной инженерии. Усилия ученых-генетиков направлены на создание и внедрение в организм больных нормального НФ1 гена, отвечающего за синтез нейрофибромина, на расшифровку и введение гена ФН2, обеспечивающего транскрипцию белка шванномина. В некоторых медицинских центрах предпринимаются попытки применения патогенетической терапии, в основе которой лежит комплексное использование стабилизаторов мембран тучных клеток, антипролиферативных препаратов и ферментов, корректирующих метаболические процессы.
Прогноз и профилактика
Нейрофиброматозы являются прогностически благоприятными заболеваниями – малигнизация опухолей происходит редко, в большинстве случаев больные остаются трудоспособными и социально адаптированными. При правильных и регулярных реабилитационных мероприятиях нарушения со стороны костной системы и задержка умственного развития успешно корректируются. Поскольку заболевание является наследственным, профилактика возможна на этапе планирования беременности, парам из групп риска (с отягощенным семейным анамнезом) рекомендуется медико-генетическое консультирование с определением вероятности рождения больного ребенка.
Нейрофиброматоз у детей
Нейрофиброматоз у детей – болезнь Реклингхаузена – это наследственное заболевание, при котором поражается кожа с образованием пигментных и опухолевидных пятен, глаза, нервная система, внутренние органы, кости ребенка. На сегодня не известны причины появления болезни. Есть данные, что болезнь имеет наследственный характер. А «спусковыми механизмами являются нейрогенный, эндокринный, дизонтогенетический факторы. В основе болезни может быть генетическая мутация, тогда ребенок рождается уже с данным диагнозом.
Нейрофибромы часто появляются в детстве, особенно в пубертатном периоде. Первый признак всегда – формирование пигментных пятен. Многие нейрофибромы можно удалить. Но 3-5% доброкачественных опухолей (по статистике) преображаются в злокачественные.
Типы нейрофиброматоза у детей:
НФ1 является более распространенным – 1 случай на 4 тыс новорожденных. Он именуется также болезнью Реклингхаузена. НФ2 бывает 1 раз на 50000 рождений. При нем обнаруживают вустороннюю акустическую нейрофиброму. В семьях, где есть несколько больных с рассматриваемым диагнозом, болезнь может проявляться в различных физических отклонениях. Осложнения могут быть также различными.
Что провоцирует / Причины Нейрофиброматоза у детей:
Нейрофиброматоз у детей почти всегда передается по наследству или является результатом мутации в организме. При наличии патологического гена у ребенка почти 100%-е шансы заболеть. Но выраженность нарушений составляет около 50%. При нейрофиброматозе у матери или отца риск ребенка заболеть составляет 50%. Если больны и мать и отец, то заболевают примерно 67 детей из 100.
Патогенез (что происходит?) во время Нейрофиброматоза у детей:
Во всех органах и тканях разрастаются доброкачественные (нераковые) опухоли, состоящие из соединительной ткани и пигментных клеток. Вокруг нервных стволов разрастаются опухоли, именуемые невриномами или неврофибромами. На коже это проявляется пигментными пятнами и образованиями, которые типичны для поражений печени (жировые бляшки, расширенные в виде кист сосуды). Это вызвано поражением печеночной ткани опухолевыми клетками.
Образования в виде пигментных пятен появляются даже на сетчатке глаз, что называется факоматозой сетчатки. Опухоли формируются на слизистой рта. Они состоят из соединительной ткани, выглядят как небольшие по размерам упругие плотные узелки. Они могут быть большими, но в любом случаев поверхности их гладкие и ровные.
Почти всегда новообразования выше уровня слизистой, могут находиться и внутри тканей, что усложняет их нахождение и диагностику. Изменения в конечностях и туловище различные. Кости могут быть слабо развиты, или же наблюдается значительное утолщение их, потому руки и ноги становятся уродливой формы, что сказывается на жизни ребенка. Искривление позвоночного столба, которое негативно отражается на функционировании спинного мозга, является стойким и постоянным изменением.
Что касается нервной системы, при нейрофиброматозе у детей снижается интеллектуальный уровень. Они впадают в тяжелую депрессию, которую крайне тяжело излечить. Патологический ген наследуется по аутосомно-доминантному признаку. Носительства не бывает. Чаще всего болеют лица мужского пола – в 2 раза чаще девочек и девушек.
Симптомы Нейрофиброматоза у детей:

Типичный симптом нейрофиброматоза у детей – пигментные пятна на спине, в паховой зоне, на внутренних поверхностях рук и ног. Они светло-кофейного оттенка, в частых случаях вокруг них есть депигментированные зоны кожи. При заболевании деформируется позвоночник, формируется сколиоз, могут быть краевые дефекты тел позвонков, их суставных и поперечных отростков, узуры нижних краёв задних отделов рёбер, расширение межпозвоночных отверстий и эрозии их краёв. Характерный симптом болезни – слоновость.
При нейрофиброматозе опухоли бывают в виде кожных разрастаний с пигментацией или узлов различной величины в мягких тканях и под кожей, что называется узловыми опухолями.
Изменения в костной ткани
При рассматриваемом диагнозе может быть дисплазия или утолщение отдельных костей, деформация костей черепа, лицевых костей, удлинение ног или рук ребенка. Также это могут быть изменения при жизни ребенка, когда на кости давят опухоли: деструкции, периостальные наслоения. Сюда же причисляют и деформацию позвоночника.
Что должно насторожить родителей?
При нейрофиброматозе, прежде всего, появляются пятна светло-коричневого оттенка. Если вы заметили у ребенка несколько таких пятен, это повод срочно обратиться к врачу. При подтверждении диагноза нужно следить за формированием опухолей и, если они появятся, срочно идти к доктору.
Обнаружение заболевания на ранних сроках очень важно для лечения. При НФ1 больше половины детей имеют легкие признаки, которые не нуждаются в лечении или требуют легкой терапии. При НФ1 у ребенка дети и в начале взросления остаются относительно здоровыми, у них почти никогда не возникает серьезных осложнений. При нейрофиброматозе второго типа ребенку нужна поддержка и лечение, чтобы вести полноценную и продуктивную жизнь.
Возрастные периоды выявления признаков нейрофиброматоза I
| Симптом | Пятна «кофе с молоком» | Диффузные плексиформные нейрофибромы | Гиперпигментация подмышечной и/или паховой зоне | Кожные нейрофибромы |
| Ранний детский возраст (0—2 года) | + | + | + | |
| Дошкольный возраст | + | |||
| Школьники и подростки (6—16 лет) | + | |||
| Взрослые (старше 16 лет) | + |
Диагностика Нейрофиброматоза у детей:
Для подтверждения диагноза «нейрофиброматоз» необходимо наличие у ребенка минимум 2 из ниже названных признаков:
Для обнаружения опухолей и диагностики проблем с костями скелета применяют такие два метода: МРТ и рентгенография. У детей с нейрофиброматозом окружность головы больше, чем у одногодок без такого диагноза. Для диагностирования НФ2 у детей нужно проверить слух. Для этого применяют аудиометрию, проводят проверку мозга, ушных нервов и спинного мозга, чтобы обнаружить возможные опухоли.
Важную информацию может дать изучение семейной истории ребенка. При наличии в ней нейрофиброматоза первого или второго типа можно провести генетическую экспертизу. Но нельзя сказать, что ее результаты точны на 100%. У новорожденного ребенка в некоторых случаях можно обнаружить отклонения при помощи биопсии хориона или амниоцентеза.
Для постановки диагноза «нейрофиброматоз первого типа» нужны консультации таких специалистов:
Врачу может понадобиться осмотр большого количества родственников, если в семье есть хотя бы один член с пятнами на коже цвета кофе с молоком, или же если на этого члена семьи указывает семейный анамнез.
Дифференциальная диагностика
Пятна цвета кофе с молоком обнаруживают не только при рассматриваемой болезни, но при более ста наследственных болезнях и синдромах. Следует отличать также нейрофиброматоз I и II типа. При втором опухоли доброкачественные, но более агрессивные, чем при первом типе.
Абсолютным диагностическим критерием НФ1 является наличие у больного двусторонних неврином VIII пары черепных нервов. При втором типе могут быть обнаружены глиомы, менингиомы, шванномы.
Требуется отличие нейрофиброматоза I от синдрома Клиппеля-Тренаунау-Вебера, синдрома Протея, рассеянного липоматоза и т.д.
Дифференциальная диагностика НФ2 у детей проводится с такими болезнями:
Нейрофиброматоз имеет общие черты с таким заболеванием как пятнистый невус, который представляет собой проявление генетического мозаицизма. Часть исследователей рассматривают пятнистый невус как сегментарный нейрофиброматоз. У детей с пятнистым невусом может развиться НФ1. Невус у ребенка бывает с рождения или же его находят у ребенка, пока ему нет 3 лет. При пятнистом невусе у ребенка есть светло-коричневые пятна, на их фоне имеются темноокрашенные вкрапления небольших размеров.
Невус у детей имеет большие размеры, но может быть небольшим. Есть случаи 2-сторонних поражений. Пятнистые невусы не сходят с тела ребенка на протяжении всей жизни. Есть совсем небольшой риск злокачественного перерождения болезни. При диагностике очень важно отличать пятнисты невус и нейрофиброматоз.
Лечение Нейрофиброматоза у детей:
Терапия НФ1 у детей заключается в удалении нейрофибром и лечении осложнений. Удаляются нейрофибромы в косметических целях. А терапия осложнений включает избавление от таких нарушений и заболеваний:
При злокачественных нейрофибромах (что бывает в 3-5 случаях из 100) необходимо хирургическое вмешательство, химиотерапия или облучение. Для лечения нейрофиброматоза 2 у детей может быть необходимо удалить опухоль слухового нерва, что иногда кончается глухотой. Слуховой аппарат в таких случаях не дает эффекта.
Управление по контролю за продуктами и лекарствами (США) в 2000 году утвердило использование слуховых имплантатов для людей с потерей слуха при нейрофиброматозе II. Такой прибор позволяет передавать звуки сразу в мозг, потому человек может слышать некоторых звуки. На сегодняшний день проводятся исследования и эксперименты с лекарственными средствами, чтобы развить другие методы лечения рассматриваемой болезни.
Прогноз при нейрофиброматозе у детей благоприятный в большинстве случаев. Как уже было отмечено, нейрофибромы перерождаются в злокачественную форму крайне редко. При распространенном поражении активность и трудоспособность ребенка резко снижена.
Профилактика Нейрофиброматоза у детей:
Специфические профилактические меры не разработаны. Рекомендуется медико-генетическое консультирование, особенно тем семьям, в которых хотя бы у одного члена есть нейрофиброматоз любого типа. Не рекомендуется рекомендуют заводить детей тем семьям, у родственников которых был диагностирован нейрофиброматоз.
К каким докторам следует обращаться если у Вас Нейрофиброматоз у детей:
Нейрофиброматоз у ребенка что это
Нейрофиброматоз 1 типа (НФ1) включает не менее 85% всех случаев нейрофиброматоза. Распространенность заболевания составляет примерно 1:3000. Нейрофиброматоз 1 типа (НФ1) наследуется по доминантному типу с пенетрантностью 98%, но с высокой степенью фенотипической вариабельности. В соответствии с публикациями Huson et al. (1988) и North (1993), приблизительно в половине случаев заболевание проявляется минимально. Передача по отцовской линии более характерна, чем по материнской. Редкие случаи мозаицизма генеративной отцовской линии могут объяснить случаи рождения более одного больного ребенка у клинически здоровых родителей (Lazaro et al., 1994).
Около трети случаев заболевания являются результатом вновь возникших мутаций; частота мутаций составляет приблизительно 1:10000 гамет на поколение.

а) Кожные проявления. Кожные проявления при нейрофиброматозе первого типа представлены аномалиями пигментации и опухолями.
Пятна цвета «кофе с молоком» являются отличительным признаком НФ1 и обнаруживаются практически во всех случаях. Их размер меняется с возрастом; часто при рождении такие пятна отсутствуют, но появляются в раннем возрасте, хотя они не так заметны в младенчестве, как позднее. Обычно к году обнаруживается не менее шести пятен диаметром 0,5-1 см, но в редких случаях их может быть меньше.
Среди 46 детей с такими пятнами, в перспективном исследовании Korf (1992), у 27 развились другие признаки НФ1 (в большинстве случаев в течение трех лет), у 6 развился сегментарный нейрофиброматоз, у трех были установлены другие диагнозы и у 8 детей диагноз не был установлен. Веснушки в подмышечной области встречались у 84% из 200 детей, наблюдаемых North (1993), а веснушки в паховой области выявлялись приблизительно у половины пациентов.
Над плексиформной нейрофибромой может наблюдаться диффузная пигментация. Реже встречаются ксантомы и ангиомы. Нейрофибромы могут располагаться внутрикожно и иметь фиолетовую окраску и мягкую консистенцию или подкожно в виде плотных опухолей, расположенных вдоль стволов периферических нервов. Размер нейрофибромы может варьировать от нескольких миллиметров до 3-4 см.
Нейрофибромы иногда обнаруживаются у детей младше 10 лет, но их количество неуклонно увеличивается с возрастом, особенно в пубертатном периоде (Riccardi, 1992). При поражении крупных нервов нейрофибромы могут приводить к периферической невропатии.
Плексиформные нейрофибромы состоят из кожных и подкожных элементов, образующих гигантские опухоли и представляют собой наиболее серьезное осложнение нейрофиброматоза 1 типа (НФ1). Данные образования могут трансформироваться во внутричерепные и интраспинальные опухоли. Плексиформные опухоли окологлазничной области, встречающиеся у 5% пациентов, могут вызывать птоз и нарушения зрения.
Крупные плексиформные нейрофибромы встречаются в 25-32% случаев (Huson et al., 1988; North, 1998), и с возрастом имеют тенденцию к увеличению размеров. Опухоли могут приводить к косметическим дефектам, а крупные образования на шее могут смещать и сдавливать сосудистые и дыхательные структуры, приводил к угрожающим жизни состояниям. Лечение нейрофибром хирургическое, но проведение его затруднено, в связи с чем изучаются другие методы (Packer and Rosser, 2002). Плексиформные нейрофибромы конечностей могут сопровождаться частичным гигантизмом.
Возможно появление опухолей лица, что может сопровождаться экзофтальмом и в редких случаях односторонней мегацефалией (Cutting et al., 2002). Подобные нарушения могут сопровождаться аномалиями костей глазницы и во избежание вторичной амблиопии требуют немедленного лечения в случае, если веко полностью закрывает глаз. Большая часть плексиформных нейрофибром образуется до двухлетнего возраста, а некоторые могут присутствовать с рождения.
Поражение радужки в виде пигментных гамартом, известное как узелки Лиша, специфично для нейрофиброматоза 1 типа (НФ1). Узелки Лиша к шести годам обнаруживаются у 22-30% пациентов, страдающих НФ 1, а к 12 годам — практически у всех пациентов (Flueler et al., 1986; North, 1998).
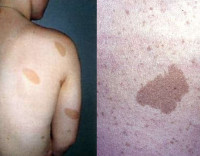
 Кожные проявления нейрофиброматоза 1 типа;
Кожные проявления нейрофиброматоза 1 типа;
на верхнем рисунке — пятна цвета «кофе с молоком», на нижнем рисунке — множественные нейрофибромы.
б) Неврологические проявления. Неврологические проявления НФ1 разнообразны и включают умственную отсталость, специфические затруднения в обучении, а также симптомы и признаки внутричерепных и внутрипозвоночных опухолей (North, 1998, 2000; Evans et al., 1999; Friedman 2002).
Макроцефалия (>98-го центиля) отмечается только у 16-45% детей, страдающих НФ1 (Huson et al, 1988, North 1997), и не коррелирует с неврологическими или нейрофизиологическими отклонениями (Cutting et al., 2002). Данное проявление связано с увеличением размеров головного мозга и не сопровождается симптомами повышения внутричерепного давления. Головные боли отмечаются приблизительно у половины пациентов и могут носить мигренеподобный характер.
Умственная отсталость и специфические затруднения в обучении относятся к хорошо известным проявлениям НФ1 (Riccardi, 1992). Задержка умственного развития встречается относительно редко: по данным Riccardi, частота данного отклонения составляет 8%, но в работах других авторов встерчается меньшая частота (North, 1997; North et al., 2002). Тем не менее, коэффициент IQ среди пациентов, страдающих НФ1, в среднем ниже, чем среди сибсов.
Специфические затруднения в обучении являются частой проблемой, затрагивающей от 32% (Hofman et al., 1994) до 65% случаев (North et al., 1998), если в качестве критерия выбирается ухудшение исполнения заданий. Наиболее вероятны затруднения при выполнении речевых заданий и высокая частота дефектов речи, но возможно и нарушение любых других когнитивных способностей (Cutting et al., 2000). Часто встречается синдром дефицита внимания без гиперактивности, который может поддаваться терапии амфетаминами.
Некоторые исследователи отмечают значимую взаимосвязь между когнитивными нарушениями и наличием участков повышенной интенсивности при проведении МРТ в Т2-режиме (Denckla et al., 1996; Wang et al., 2000; Goh et al., 2004), но другими авторами такой взаимосвязи выявлено не было (Moore et al., 1996; Rosenbaum et al., 1999), что свидетельствует о необходимости дальнейшего изучения данной взаимосвязи. Часто отмечается нарушение социальных навыков, приводящее к затруднению в общении (Barton and North 2004). Так же часто встречаются изменения поведения (Kayl и Moore, 2000).
Среди детей, страдающих НФ1, эпилепсия встречается чаще, чем среди населения в целом, по имеющимся данным частота данного заболевания колеблется между 3,5% (North, 1998), 4,2% (Kulkantrakorn и Geller, 1998) и 7,3% (Huson et al., 1988). Возможно развитие всех типов судорог, включая инфантильные спазмы (Motte et al., 1993; Fois et al, 1994).
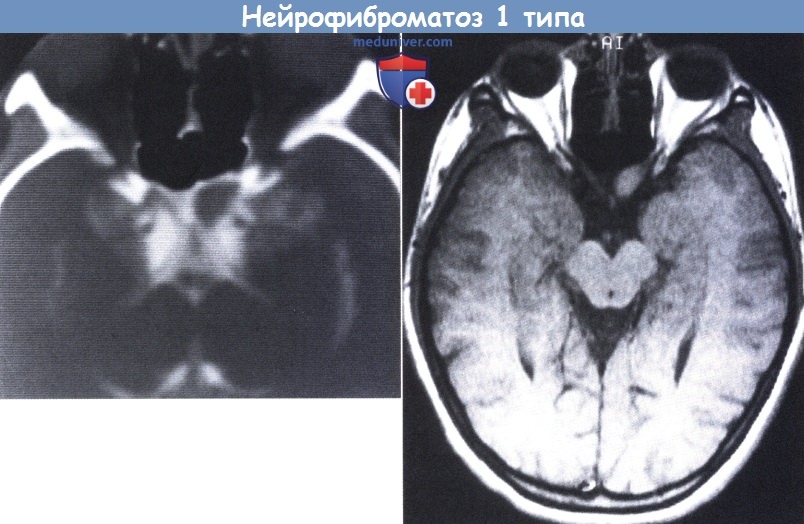 НФ1 у 12-летней девочки. На верхнем рисунке: на компьютерной томограмме, проведенной после люмбального введения контрастного вещества (метризамида), видно небольшое образование в месте начала левого зрительного нерва.
НФ1 у 12-летней девочки. На верхнем рисунке: на компьютерной томограмме, проведенной после люмбального введения контрастного вещества (метризамида), видно небольшое образование в месте начала левого зрительного нерва.
На нижнем рисунке: на снимке МРТ в Т2-режиме ясно видна небольшая глиома зрительного нерва.
Глиомы зрительного нерва встречаются у 15-20% пациентов, страдающих НФ1 (North, 1998). Гистологически данные образования представляют собой типичные полицитарные астроцитомы, но могут отличаться от других глиом зрительного нерва наличием арахноидального глиоматоза, окружающего зрительный нерв (Seiff et al., 1987). Глиомы могут ограничиваться поражением зрительного нерва или распространяться на хиазму и ретрохиазмальный участок зрительного пути. От 10% до 60% глиом зрительного нерва связаны с НФ1. Отмечается тенденция к увеличению относительного числа глиом зрительного нерва, в особенности двусторонних глиом, которые обнаруживаются практически только среди пациентов, страдающих НФ1.
Клинические проявления включают проптоз и снижение остроты зрения, но в настоящее время глиомы часто остаются бессимптомными и обнаруживаются при систематическом рентгеновском обследовании пациентов с НФ1. До 39% случаев заболевания проявляется преждевременным половым созреванием (Habiby et al., 1995). Доля симптоматических случаев составляет 20-30%; симптомы заболевания чаще всего манифестируют в раннем возрасте (до 6 лет, а часто к двум годам) (Listernick et al., 1994). Глиомы зрительного нерва при НФ1 можно разделить на две группы: стабильные и активно растущие, угрожающие зрению.
Только у 52% пациентов, у которых глиомы зрительного нерва выявлялись при проведении МРТ, отмечалось развитие симптомов (Listernick et al., 1997). Появление симптомов в возрасте старше шести лет, а также прогрессирование заболевания после 10 лет встречается крайне редко, тем не менее, зарегистрированы случаи позднего проявления и прогрессирования заболевания (Listernick et al., 2004). Даже в случае форм, сопровождающихся клиническими проявлениями, исход заболевания зачастую носит благоприятный характер, что следует принять во внимание при назначении лечения и проведении повторной диагностической визуализации. Из 104 пациентов (в том числе 88 детей), страдающих опухолями центральной нервной системы, включая 88 случаев глиом зрительного нерва, 55 из которых сопровождались клиническими проявлениями, у большинства отмечался благоприятный исход (Guillamo et al., 2003).
Высокая частота случаев заболевания, сопровождающихся клиническими проявлениями, объясняется тем, что исследование проводилось в области хирургии. Внутриглазничные глиомы редко распространяются внутрь черепа.
Развитие глиом зрительного нерва при НФ1 носит доброкачественный характер чаще, чем при изолированных формах заболевания (Listernick et al., 1994), но единого мнения по данному вопросу не существует. Опухоли, располагающееся спереди от хиазмы, часто являются стабильными, в то время как глиомы, расположенные позади хиазмы, часто склонны к инвазивному росту. Наличие глиомы зрительного нерва может быть предположено при обнаружении увеличенного глазничного отверстия, но данный признак не является специфичным, и удлинение зрительного нерва и расширение твердой мозговой оболочки может походить на опухоль (Riccardi, 1992; North, 1997).
Современные методы визуализации нервной системы изменили методику диагностики опухолей зрительного нерва среди пациентов, страдающих НФ1. На снимке, полученном методом КТ, опухоль выглядит как неравномерное утолщение, которое может распространяться на хиазму с выпячиванием или заполнением цистерны хиазмы. В настоящее время продолжается спор о показаниях для проведения визуализации при НФ1. По общему мнению, визуализация необходима в случае клинического проявления заболевания. В случае бессимптомного течения заболевания по решению, принятому на конференции Национального института здоровья (1988 г.) не рекомендуется систематическое обследование, так как лечение обычно не назначается.
Riccardi (1992 г.) придерживается противоположного мнения, так как некоторые агрессивные глиомы могут прогрессировать бессимптомно в течение относительно длительного времени и в дальнейшем приводить к повреждениям, которых можно было бы избежать при ранней диагностике. Регистрация вызванных зрительных потенциалов может обеспечить раннюю диагностику глиомы, но при этом велика доля ложноположительных результатов; при использовании данного метода часто выявляется распространение глиомы на зрительный тракт (Di Mario et al., 1993).
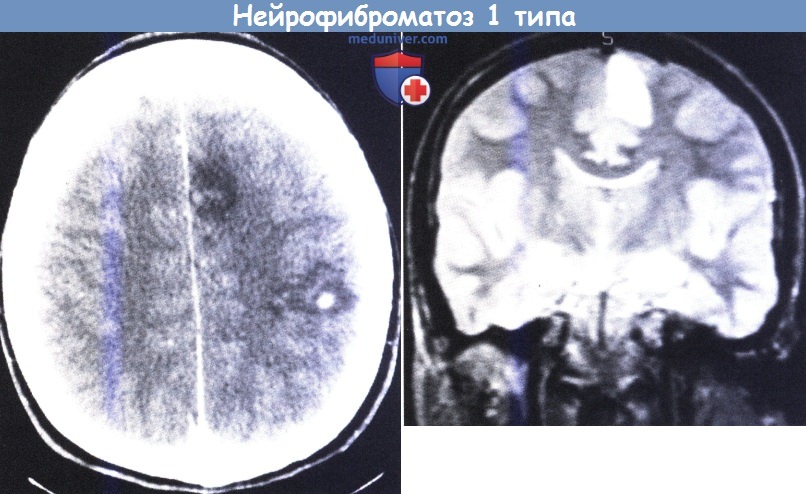 10-летняя девочка с НФ1, сопровождающимся недавно начавшимися правосторонними парциальными моторными припадками.
10-летняя девочка с НФ1, сопровождающимся недавно начавшимися правосторонними парциальными моторными припадками.
На верхнем рисунке: на КТ видны области пониженной плотности, расположенные в левом полушарии, с повышенным контрастированием в центре задней части.
На нижнем рисунке: на снимке МРТ в Т2-режиме (коронарный срез) видно усиление сигнала в переднемедиальной области слева.
В дальнейшем было удалено две опухоли (астроцитомы HI степени) с полным устранением припадков на один год.
В настоящее время не существует единого мнения по поводу лечения глиомы зрительного нерва (Riccardi, 1992). Большая часть опухолей, характеризующихся стабильностью, в особенности расположенных в передней части зрительного пути, требует тщательного наблюдения без проведения хирургического или любого другого вмешательства. В рамках совместного исследования распространение опухоли на хиазму зрительного нерва отмечалось только в четырех из 106 случаев односторонней опухоли (Wilson, 1998). В случае растущей глиомы зрительного нерва, приводящей к заметному проптозу и полной односторонней потере зрения, хирургическое удаление приводит к удовлетворительным результатам.
В случае нестабильных нарушений эффективна радиотерапия. Тем не менее, обычно используемая доза облучения (52 Грея) в трети случаев приводит к задержке умственного развития и изменениям со стороны эндокринной системы у детей. Среди детей младше пяти лет, у которых частота такой реакции на облучение достаточно высока, лучевая терапия обычно заменяется химиотерапией. Часто применяется комбинация препаратов с подтвержденной эффективностью: винкристин в сочетании с карбиплатином (North, 1997).
15 пациентов были живы при последующем наблюдении через несколько лет, несмотря на то, что за исключением шунтирования (в случае необходимости) лечения не проводилось. Авторами исследования сделан вывод о том, что данные опухоли занимают промежуточное положение между гамартомами и классическими опухолями ствола мозга, и до появления признаков прогрессирования опухоли следует воздерживаться от инвазивных методов лечения. Сходные выводы были сделаны по результатам исследования 21 пациента (Pollack et al., 1996). Есть сообщение (Schmandt et al., 2000) о спонтанном регрессе некоторых астроцитом.
Нередко встречаются множественные опухоли мозга (Hochstrasser et al., 1988). Внутричерепная кальцификация, затрагивающая центральные ядра или перивентрикулярное пространство, встречается редко (Arts and Van Dongen, 1986) и имеет неопухолевое происхождение. Опухоли периферических нервов у детей встречаются редко, но могут иметь злокачественный характер (Drouet et al., 2004).
Внутрипозвоночные опухоли встречаются редко и представлены в основном астроцитомами. Внутрипозвоночные нейрофибромы обнаруживаются преимущественно в шейно-грудном отделе. Данные опухоли встречаются нередко и могут располагаться как вне, так и внутри позвоночника и составлять единое целое с подкожными плексиформными нейрофибромами. Интрамедуллярные опухоли, такие как эпендимомы и астроцитомы не являются признаком НФ1. Имеются данные о редких случаях нейрофиброматоза позвоночника с множественными симметричными опухолями (Pascual-Castroviejo et al., 2007).
Гамартомы головного мозга и мозжечка, так называемые «неидентифицируемые светлые образования», представляют собой участки повышенной интенсивности, выявляемые методом МРТ (в Т2-режиме), располагающиеся чаще всего в таламусе, ножках мозжечка и полушариях, а также в белом веществе полушарий в зрительном тракте и лучистости. Значимость описанных изменений полностью не ясна. Описанные изменения в большинстве случаев не соответствуют области распространения опухоли и могут оставаться стабильными или исчезать. Такие образования часто встречаются как среди пациентов, не страдающих глиомами зрительного нерва, так и в сочетании с данными опухолями (Duffner et al., 1989, North 1998).
Области повышенной интенсивности выявлялись у 16 детей при проведении МРТ в Т1-режиме и обычно соответствовали аномалиям, выявленным в Т2-режиме. В связи с совершенствованием технологии МРТ частота данных аномалий последнее время увеличилась, и некоторые авторы предлагают использовать описанные изменения в качестве критерия диагностики НФ1 (Curless et al, 1998, DeBella et al., 2000). Описанные изменения присутствовали в 25 из 29 случаев (Rosenbaum et al., 1999) и в 89% случаев (Raininko et al., 2001).
В ходе более поздних исследований проводилось детальное изучение данных областей. В рамках описанных исследований данные изменения чаще выявлялись в режиме протонной плотности (80%), чем в Т2-режиме (50%); изредка отмечалось усиление данных изменений при применении гадолиния.
У пяти пациентов измененные участки увеличивались в размере, в дальнейшем в четырех случаях отмечалась регрессия и исчезновение изменений. Данные изменения могут меняться с развитием, имеют тенденцию к исчезновению с возрастом, но могут и увеличиваться во взрослом возрасте, также были зарегистрированы случаи двухфазного развития (Kraut et al., 2004). Данные образования редко встречаются при наличии опухоли зрительного пути.
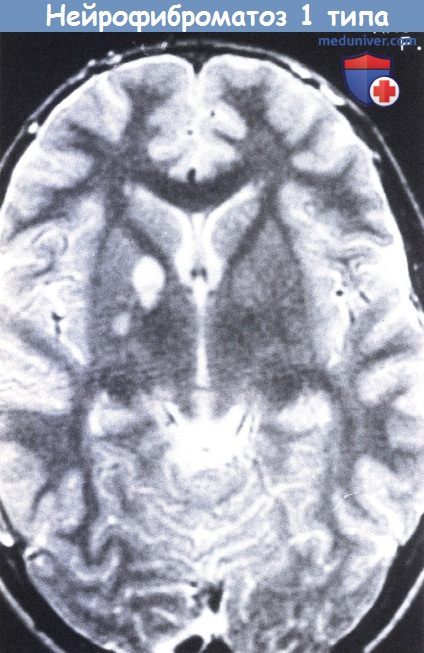 НФ1 у 14-летней девочки.
НФ1 у 14-летней девочки.
При проведении МРТ в Т2-режиме выявлены области усиления сигнала («гамартомы») в правом базальном ганглии и таламусе.
Области повышенной интенсивности имеют склонность к обратному развитию и часто исчезают до наступления совершеннолетия (Sevick et al., 1992). Гистологическая природа данных образований неизвестна. У детей и подростков они встречаются чаще, чем у взрослых. Размеры изменений зрительного тракта и бледного шара больше, чем при иной локализации, являясь источником слабого аномального сигнала при проведении МРТ в Т2-режиме (Inoue et al., 1997). В редких случаях аномальная интенсивность в Т2-режиме МРТ соответствовала дисплазии глиальных клеток или губчатым изменениям, а не гамартомам (Di Paolo et al., 1995).
При проведении МРТ в протонном режиме в описанных областях изначально отмечалось повышение уровня холина и относительно нормальное содержание N-ацетиласпартата с последующим снижением его уровня, что позволяет предположить вторичную гибель нейронов.
Пороки развития центральной нервной системы, в особенности патология миграции нейронов, при НФ1 встречаются нечасто. Тем не менее, зарегистрированы случаи гемимегалэнцефалии (Cusmai et al., 1990). Встречаются опухоли периферических нервов. Зарегистрирован один случай смерти вследствие нейрофибромы блуждающего нерва (Chow et al., 1993).
Гидроцефалия в большинстве случаев является следствием стеноза сильвиева водопровода (Afifi et al., 1988, Riviello et al., 1988). Данная патология обычно развивается медленно и выявляется поздно. Гидроцефалия чаще всего связана с диффузным или мембранозным глиозом водопровода мозга. Причиной гидроцефалии могут быть небольшие опухоли в области ножек мозга, поэтому МРТ в сагиттальной проекции с использованием Т1- и Т2-режимов обязательна во всех случаях. Интенсивный сигнал на снимках, выполненных в Т2-режиме, выявлен в 7 из 9 случаев (Pou-Serradell and Ugarte-Elola 1989, Valentini et al, 1995). Другие случаи увеличения желудочков среди пациентов, страдающих НФ1, были связаны с мальформацией Киари 1 типа (Afifi et al., 1988) и опухолями задней черепной ямки.
Аномалии черепа часто встречаются при НФ1. Черепно-лицевая дисплазия может затрагивать любую часть свода черепа, но чаще всего отмечается в затылочной части в области ламбдовидного шва. Типична локализация дисплазии в области костей, участвующих в образовании глазницы, в особенности большого крыла клиновидной кости. Большое крыло частично отсутствует, что в случае большой площади дефекта приводит к образованию пульсирующего экзофтальма. Часто дисплазия затрагивает малое крыло и турецкое седло. Эктазии твердой мозговой оболочки могут приводить к двустороннему увеличению наружного слухового прохода, что может свидетельствовать о НФ2, но не сопровождается шванномами VII пары черепных нервов (Inoue et al., 1997). Дефекты свода черепа в областях, прилежащих к лямбдовидному шву, не сопровождаются клиническими симптомами.
Сколиоз встречается в 20% случаев и может быть достаточно выражен, что требует хирургического лечения. Сколиоз изредка сопровождается околопозвоночными нейрофибромами. Часто встречаются аномалии позвонков, а неровности тел позвонков в качестве проявления сколиоза являются частой находкой при рентгенологическом обследовании при НФ1. Боковые выпячивания истонченной твердой оболочки через межпозвонковые отверстия (боковые грыжи мозговых оболочек) являются редким осложнением НФ1 (Riccardi, 1992). Часто встречается псевдоартроз большеберцовой кости (North, 1997).
в) Другие осложнения нейрофиброматоза первого типа (НФ1). Другие осложнения НФ1 включают задержку роста, зарегистрированную примерно у 25% пациентов (North, 1998). У 122 из 251 ребенка рост был ниже 25-го перцентиля, а у 68 — ниже 10-го перцентиля (Vassilopoulou-Sellin et al., 2000). Типичны заболевания глаз, в особенности глаукома и гидрофтальм, костная дисплазия и кисты паутинной оболочки; аномалии электроэнцефалограммы (ЭЭГ) встречаются у 15% пациентов (Riccardi, 1992).
Проявления со стороны сосудов (Tomsik et al, 1976) включают гипертензию в сочетании со стенозом почечной артерии или без него (обязательно обследование для выявления данной патологии) и (в редких случаях) острое нарушение мозгового кровообращения (Rosser et al., 2005). Зарегистрированы случаи множественного стеноза сонных артерий с развитием болезни мойя-мойя (Rizzo and Lessell, 1994). У 13 из 69 детей, после лучевой терапии по поводу глиомы хиазмы зрительных нервов, развился стеноз внутричерепной части сонных артерий (Grill et al., 1999). Частота злокачественных заболеваний, самым частым из которых является фибросаркома, среди пациентов, с НФ1 выше, чем среди населения в целом, но редки у детей.
Отмечены случаи лейкемии (Clark and Hutter 1982), в особенности миелоидного происхождения, которая может сопровождаться ксантомами кожи. Опухоли висцеральных и эндокринных органов часто встречаются в старческом возрасте (Huson et al., 1988). Могут встречаться независимые висцеральные опухоли автономной нервной системы, в частности ганглионейрофиброматоз кишечника с обструкцией просвета кишки (Kim and Kim, 1998).
Редактор: Искандер Милевски. Дата публикации: 30.11.2018