Нейрональный цероидный липофусциноз 2 типа что это
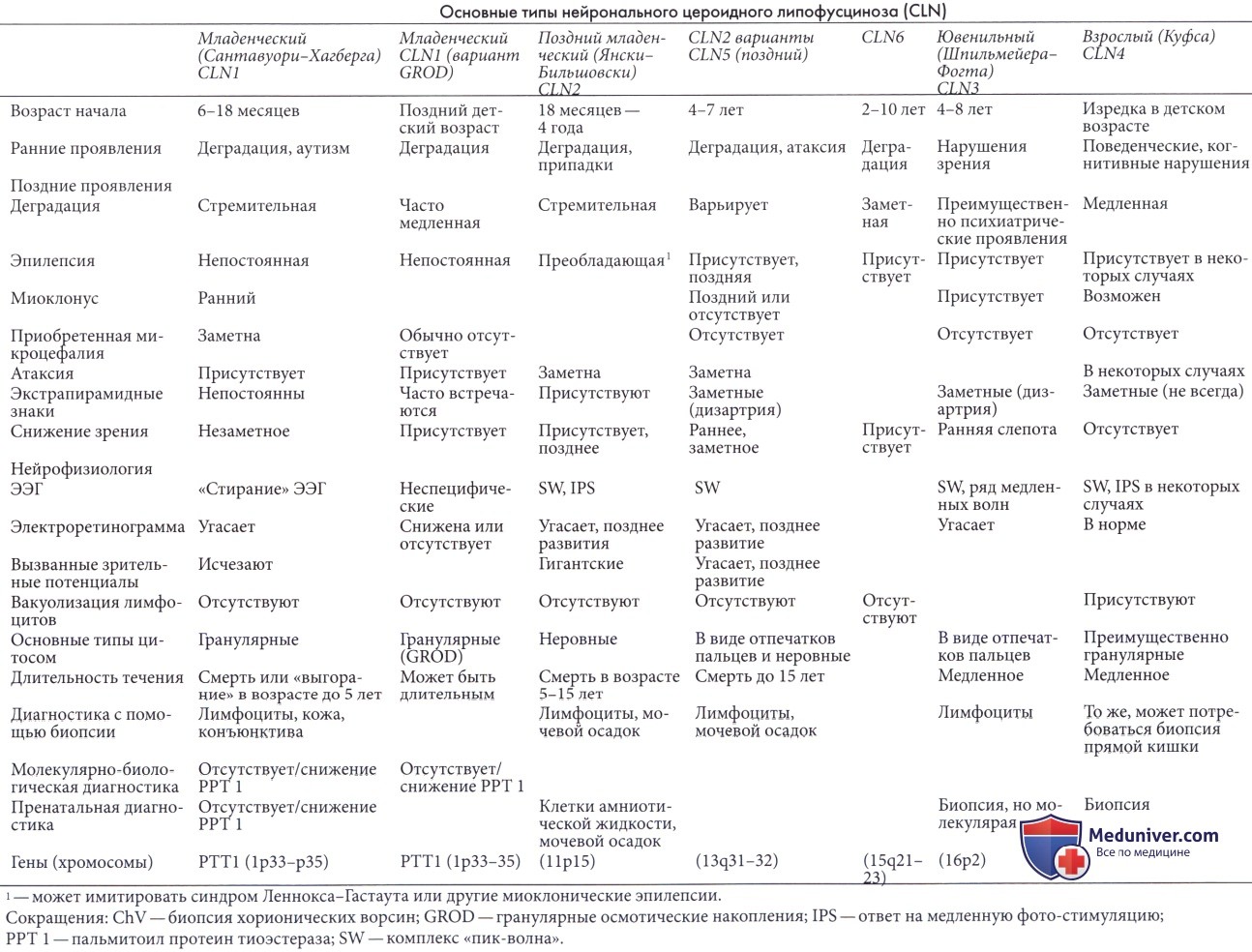
а) Нейрональный цероидный липофусциноз взрослых (болезнь Куфса, CLN4). Несмотря на название, CLN взрослых может начинаться в детстве или подростковом возрасте, первые симптомы могут появляться в возрасте трех лет (Berkovic et al., 1988). Заболевание начинается незаметно и проявляется преимущественно когнитивными и поведенческими нарушениями. В конечном счете формируется слабо выраженная деменция. При длительном течении заболевания могут появиться экстрапирамидные признаки, в частности, дискинезия лица.
Аномалии зрения или ухудшение зрения не отмечаются. У некоторых пациентов могут развиваться миоклонические припадки. Выделяют два типа заболевания (Berkovic et al., 1988): с прогрессирующей миоклонической эпилепсией и нейропсихиатрическими изменениями (Sadzot et al., 2000), с деменцией и двигательными, мозжечковыми и экстрапирамидными нарушениями. На ЭЭГ может выявляться заметная реакция на фотостимуляцию с высокоамплитудными пиками, синхронными со вспышками при низкой частоте повторения. Большая часть зарегистрированных случаев являлись спорадическими, вероятно, аутосомно-рецессивными. В редких семьях вероятно доминантное наследование.
Могут обнаруживаться цитосомы, содержащие гранулярные осмиофильные включения, но обнаружение их вне нервной ткани может быть затруднено.
б) Другие атипичные формы. Такие формы могут составлять до 10-20% случаев по результатам некоторых наблюдений (Dyken и Wisniewski 1995; Goebel и Wisniewski 2004). Предложено несколько классификаций, некоторые содержат более 15 различных групп, но ни одна из классификаций полностью не признана (Dyken и Wisniewski, 1995). Предстоит еще установить, являются ли редко встречающиеся формы отдельными нозологическими единицами или только вариантами течения классических форм заболевания.
Несколько случаев врожденного CLN (тип Нормана-Вуда) были зарегистрированы Dyken and Wisniewski (1995). Данная форма характеризуется стремительным развитием смертельной дыхательной недостаточности и эпилептического статуса; недавно было высказано предположение, что причиной заболевания является дефицит лизосомального катепсина D (Siintola et al., 2006). В начале заболевания появляются выраженные симптомы со стороны зрения и медленно прогрессирующие припадки и деменция. Преобладание нелинейных включений и нарушение зрения отличает данное заболевание от взрослой формы.
Пигментный вариант (Goebel et al., 1995) начинается в детстве и также характеризуется длительным течением, но отличается внешним видом накапливающихся включений. Описан хронический младенческий тип (Dyken и Wisniewski, 1995) и случаи заболевания в детстве, изолированные случаи с хореей, спиноцеребеллярными симптомами, нейропатией и медленным прогрессированием. Зарегистрировано несколько случаев, сочетающихся с остеопорозом (Takahashi et al., 1990), которые могут начинаться в пренатальном периоде.
в) Диагностика. Диагностика CLN может быть затруднена при менее типичных формах заболевания. Нейрофизиологические проявления, особенно аномалии электроретинограммы и характерный ответ на ЭЭГ при низкочастотной световой стимуляции (1-3 Гц), типичны при поздней младенческой форме и некоторых «взрослых» формах. Атрофия головного мозга, выявляемая при нейровизуализации, обычно проявляется рано, но может и не обнаружиться в первый год болезни. Снижение сигнала таламуса в Т2-режиме, выявляемое при CLN1,2, 3,4, 5, 6 и турецком типе позднего младенческого CLN, подозрительно, но не специфично (Autti et al., 2007).
Молекулярное генетическое тестирование при классических типах CLN (1, 2 и 3) путем оценки содержания пальмитоил-протеин тиоэстеразы при CLN1 и активности трипептил-пептидазы 1 при CLN2 (Young et al., 2001) является наиболее эффективным методом диагностики. Анализ мутаций возможен при формах заболевания, для которых имеется клонированный ген (Zhong, 2001). Методы диагностики редких форм заболевания до сих пор разрабатываются. При некоторых формах заболевания единственным доступным методом диагностики является выявление характерных включений в лимфоцитах, коже, конъюнктиве или других экстраневральных тканях.
Ни один из типов включений не является специфичным для конкретной формы заболевания, тем не менее, обычно имеется преобладающий тип. Результаты биопсии экстраневральных тканей изредка отказываются отрицательными. В таких случаях при биопсии прямой кишки можно выявить нейроны, содержание патологические включения.
Пренатальная диагностика форм заболевания, для которых выявлен конкретный ген, может проводиться на основании тех же молекулярных технологий. Также возможна диагностика на основании морфологической оценки включений в клетках амниотической жидкости или биоптатов ворсин хориона (Chow et al., 1993) или на основании выявления субъединицы С митохондриальной АТФ-азы при CLN2 и 3 в сочетании со сцеплением ДНК при CLN 1 и 3 (Goebel et al., 1995). В отдельных случаях CLN 1 возможно выявление носителей.
г) Лечение. Предложено лечение антиоксидантами (витамином Е и С, селеном) (Santavuori et al., 1985), но результаты, к сожалению, ограничены. При большинстве типов заболевания необходимо противосудорожное лечение. Часто эффективны вальпроат натрия и/или клоназепам, но противосудорожное лечение должно подбираться индивидуально, а припадки могут быть полностью рефрактерными.
Пациентам и их семьям необходим поддерживающий уход, чтобы помочь в борьбе с тяжким заболеванием.
Редактор: Искандер Милевски. Дата публикации: 18.12.2018
Нейрональный цероидный липофусциноз 2 типа что это
Нейрональные цероидные липофусцинозы (CLN, болезнь Баттена) представляют собой гетерогенную группу заболеваний, характеризующихся накоплением определенных липопигментов, обеспечивающих сходные морфологические свойства и окрашивание липофусцином, аутофлуоресцентным пигментом, обнаруживаемым во многих тканях животных. Несмотря на то, что термин «липофусцин», обозначающий пигмент, накапливающийся в нейронах с возрастом, используется часто, липопигменты, являющиеся компонентами накапливаемой субстанции, называемой восковидной, варьируют (Seehafer и Pearce, 2006).
Ультраструктурное строение восковидной субстанции неоднородно. Химическая природа данных пигментов описана не полностью. Липофусцин в норме накапливается в нервных клетках с возрастом, количество его варьирует и рассматривается как вещество, свойственное старению.
Основной компонент пигментных накоплений младенческого и ювенильного восковидного липофусциноза включает субъединица С АТФ-синтазы дыхательной цепи. Восковидная субстанция накапливается в основном в структурах, подобных лизосомах, характеризующихся положительной реакцией на кислую фосфатазу.
Нейрональные цероидные липофусцинозы (CLN, болезнь Баттена) являются наиболее частными дегенеративными заболеваниями детского возраста. Описано восемь основных типов и ряд недостаточно изученных форм. В настоящее время выделено и описано не менее шести различных генов, расположенных на различных хромосомах (Mole et al., 2004, 2005).
Восковидный липофусциноз является генетически детерминированным прогрессирующим дегенеративным заболеванием, поражающим ЦНС и сетчатку. Заболевания детского возраста являются аутосомно-рецессивными. Основные неврологические проявления, эпилепсия, миоклонус и когнитивная деградация отражают преимущественное поражение серого вещества (Santavuori et al., 2001).
Нейрональные цероидные липофусцинозы (CLN, болезнь Баттена) встречаются в разных этнических группах; тем не менее, отмечаются этнические вариации.
Младенческая форма, редко встречающаяся в большинстве стран, часто отмечается в Финляндии, где ее частота составляет 7,7 на 100000 живых новорожденных, в то время как ювенильная форма типична для Великобритании и Германии, другие типы заболевания также выявляются преимущественно в отдельных популяциях (Goebel и Wisniewski 2004; Mole, 2004).
Цероидный липофусциноз может рассматриваться как лизосомальное заболевание. Фактически, дефицит лизосомальных ферментов выявлен при некоторых формах (см. далее) заболевания, но аномалии и дисфункция структурных белков также участвует в патогенезе (Goebel и Winiewski, 2004).
Термин нейрональный восковидный липофусциноз не полностью отражает природу заболевания, так как липопигмент также обнаруживается во многих тканях вне нервной системы, нахождение пигмента в нервной ткани до сих пор является основой диагностики CLN. Тем не менее, выявление нескольких генетических мутаций в настоящее время позволяет осуществлять молекулярную диагностику нескольких форм заболевания.
Классификация нейрональных цероидных липофусцинозов (CLN, болезни Баттена) включает четыре основных формы: CLN1 типа или классический младенческий тип, CLN2 или поздний младенческий тип, CLN3 или ювенильный тип и CLN4 или взрослый тип (Goebel et al., 1995). В одном из исследований из 520 случаев не было классифицировано 20% (Goebel и Wisniewski, 2004).
Редактор: Искандер Милевски. Дата публикации: 17.12.2018
Нейрональный цероидный липофусциноз 2 типа что это
НЕЙРОНАЛЬНЫЙ ЦЕРОИДНЫЙ ЛИПОФУСЦИНОЗ 2 ТИП.
(НЦЛ 2, НЕДОСТАТОЧНОСТЬ ТРИПЕПТИДИЛ ПЕПТИДАЗЫ 1, КЛАССИЧЕСКИЙ ВАРИАНТ ПОЗДНЕЙ МЛАДЕНЧЕСКОЙ ФОРМЫ НЦЛ 2 ТИПА, БОЛЕЗНЬ ЯНСКОГО-БИЛЬШОВСКОГО, сLINCL).
CLN 2; CEROID LIPOFUSCINOSIS, NEURONAL, 2; CLN2; CEROID LIPOFUSCINOSIS, NEURONAL, 2, VARIABLE AGE AT ONSETJAN SKY-BIELSCHOWSKY DISEASE; NEURONAL CEROID LIPOFUSCINOSIS, LATE INFANTILE, INCLUDED; LINCL, INCLUDED
Генетика : мутации гена трипептидил пептидазы 1 (TPP1 MIM *607998). Ген картирован на коротком плече 11 хромосомы (локус 11p15).
Эпидемиология :суммарная частота встречаемости всех форм НЦЛ в мире составляет 1: 25000.
Тип наследования :аутосомно-рецессивный
Патогенез :при всех формах НЦЛ происходит накопление в лизосомах клеток аутофлюоресцентного липопигмента, который состоит из белков сапозинов А и D и/или субединицы с митохондриальной АТФ синтазы. Накапливаемый материал характеризуется аутофлюоресценцией в зелено-желтом спектре при возбуждении светом длиной от 340 до 360 нм. При гистохимическом окрашивании материал дает положительную реакцию на кислую фосфатазу и окрашивается суданом черным В, что свидетельствует о присутствии фосфолипидов. Он также является ШИК- положительным, что может указывать на высокое содержание углеводов. Большинство из этих свойств идентично свойствам так называемых пигментов «изнашивания», или «старения», обычно называемых липофусцинами или цероидами. При электронной микроскопии нейрональных и экстранейрональных тканей обнаруживают цитосомы, заполненные агрегатами характерной морфологии, описываемые как «криволинейные» профили, по типу «отпечатков пальцев», «прямолинейные» профили, гранулярные осмиофильные отложения.
Клинические проявления :в зависимости от возраста манифестации НЦЛ 2 типа подразделяют на три формы: врожденную, позднюю младенческую, юношескую формы. Классическая форма поздней младенческой формы НЦЛ широко распространена во всем мире, но с наибольшей частотой в западной Финляндии. Обычно первые симптомы появляются в возрасте от двух до четырех лет. Манифестными симптомами являются генерализованные тонико-клонические приступы, задержка речевого развития, атаксия. По мере развития заболевания присоединяются другие типы эпилептических приступов (миоклонические, парциальные, диалептические). Вслед за развитием эпилептических приступов развиваются интеллектуальные нарушения, и происходит утрата ранее приобретенных двигательных навыков. Часто у пациентов наблюдаются атаксия, мышечная гипотония, дизартрия. Зрительные нарушения, как правило, присоединяются позднее и прогрессируют очень медленно. Мышечная гипотония сменяется спастичностью, формируются сгибательные контрактуры конечностей, формируется псевдобульбарный синдром. В терминальную стадию болезни те же симптомы, что и при младенческой
Диагностика :основными методами подтверждения диагноза НЦЛ 2 типа являются определение активности ферментов в лейкоцитах крови или культуре клеток кожных фибробластов, молекулярно-генетические наследования, исследования биоптатов при электронной микроскопии
Нейрональный цероидный липофусциноз
Нейрональный цероидный липофусциноз (НЦЛ) – это группа генетических заболеваний, в основе которых лежит накопление в клеточных структурах нейронов и других тканей токсического пигмента – липофусцина. Патология наследуется по аутосомно-рецессивному типу.
Патогенез нейронального цероидного липофусциноза
В основе патогенеза лежит нарушение утилизации пигмента липофусцина. Он накапливается в тканях организма человека и в норме, но гораздо медленнее. В случае нейронального цероидного липофусциноза это накопление происходит стремительно и приводит к атрофии тканей. Липопигменты локализуются в клеточных органеллах – лизосомах, выполняющих функцию утилизации отработанных клеточных элементов.
Ранее в основе классификации нейронального цероидного липофусциноза лежал возраст пациента, в котором начинало манифестировать заболевание. В зависимости от этого выделяли 4 типа патологии:
По мере изучения наследственных болезней были выявлены мутации в генах NCL, которые и легли в основу современной классификации. В настоящее время выделяют 10 типов заболевания в зависимости от имеющихся генетических нарушений.
Клинические проявления нейронального цероидного липофусциноза
Нейрональный цероидный липофусциноз является прогрессирующим заболеванием детей и подростков, однако некоторые формы патологии могут развиваться и у взрослых людей младше 40 лет. До появления клинических проявлений дети ничем не отличаются от своих сверстников.
Манифестация заболевания проявляется одним из трех признаков:
По мере прогрессирования присоединяются следующие признаки:
Со времени возникновения первых симптомов состояние ребенка быстро ухудшается. Гибель происходит в течение нескольких лет. При благоприятных формах нейронального цероидного липофусциноза пациенты редко переживают сорокалетний рубеж.
Методы лечения нейронального цероидного липофусциноза
На сегодняшний день эффективного патогенетического лечения нет, однако состояние пациентов можно облегчить с помощью симптоматической терапии. Это назначение противоэпилептических препаратов, миорелаксантов, седативных, антипсихотических, обезболивающих средств, витаминов. Иногда временное улучшение приносит физиотерапия.
В некоторых странах ведется активный поиск методов лечения, который сосредоточен на трех направлениях:
В целом есть обнадеживающие результаты, позволяющие надеяться на победу страшной болезни в будущем. В США и Европе зарегистрирована фермент-заместительная терапия рекомбинантной альфа-церлипеназой (Бринейра). Препарат позволяет прекратить прогрессирование заболевания.
В рамках диагностического поиска используется нейровизуализация (МРТ, ПЭТ), ЭЭГ, электроретинограмма. Активно применяется гистологическое исследование, но наиболее ценной информацией обладает молекулярно-генетическая диагностика для определения мутаций в генах. Исследуется материал от обоих родителей и ребенка. На основании полученных данных делается заключение о наличии или отсутствии у пациента нейронального цероидного липофусциноза.
Публикации в СМИ
Липофусцинозы
Липофусцинозы восковидные (цероидные, цероид-липофусциноз, липофусцинозы нейрональные) — группа наследственных аутосомно-рецессивных заболеваний, характеризующихся накоплением пигментов • Хроническая ювенильная (204200, болезнь Баттена) — медленное прогрессирование поведенческих и зрительных симптомов •
Острая поздняя детская (204500, болезнь Янского–Бильшовского) • Хроническая взрослая (болезнь Куфса) • Острая детская (256730, болезнь Santavuori–Haltia) — быстрое развитие двигательных и умственных расстройств в сочетании с миоклоническими судорогами и прогрессирующей слепотой • Описано множество более редких форм.
Лечение • Симптоматическое • Реабилитационные мероприятия.
МКБ-10 • E75.4 Липофусциноз нейронов.
Код вставки на сайт
Липофусцинозы
Липофусцинозы восковидные (цероидные, цероид-липофусциноз, липофусцинозы нейрональные) — группа наследственных аутосомно-рецессивных заболеваний, характеризующихся накоплением пигментов • Хроническая ювенильная (204200, болезнь Баттена) — медленное прогрессирование поведенческих и зрительных симптомов •
Острая поздняя детская (204500, болезнь Янского–Бильшовского) • Хроническая взрослая (болезнь Куфса) • Острая детская (256730, болезнь Santavuori–Haltia) — быстрое развитие двигательных и умственных расстройств в сочетании с миоклоническими судорогами и прогрессирующей слепотой • Описано множество более редких форм.
Лечение • Симптоматическое • Реабилитационные мероприятия.
МКБ-10 • E75.4 Липофусциноз нейронов.