Гипогонадизм
Гипогонадизм – синдром, сопровождающийся недостаточностью функций половых желез и нарушением синтеза половых гормонов. Гипогонадизм, как правило, сопровождается недоразвитием наружных или внутренних половых органов, вторичных половых признаков, расстройством жирового и белкового обмена (ожирением или кахексией, изменениями костной системы, сердечно-сосудистыми нарушениями). Диагностика и терапия гипогонадизма осуществляется совместной работой эндокринологов, гинекологов и гинекологов-эндокринологов (у женщин), андрологов (у мужчин). Основу лечения гипогонадизма составляет заместительная гормональная терапия. При необходимости проводится хирургическая коррекция, пластика и протезирование половых органов.
Общие сведения
Гипогонадизм – синдром, сопровождающийся недостаточностью функций половых желез и нарушением синтеза половых гормонов. Гипогонадизм, как правило, сопровождается недоразвитием наружных или внутренних половых органов, вторичных половых признаков, расстройством жирового и белкового обмена (ожирением или кахексией, изменениями костной системы, сердечно-сосудистыми нарушениями). Различают мужской и женский гипогонадизм.
Гипогонадизм у мужчин
Классификация гипогонадизма у мужчин
Гипогонадизм делится на первичный и вторичный. Первичный гипогонадизм вызван нарушением функции тестикулярной ткани вследствие дефекта самих яичек. Хромосомные нарушения могут приводить к аплазии или гипоплазии тестикулярной ткани, что проявляется отсутствием секреции андрогенов или их недостаточностью для нормального формирования половых органов и вторичных половых признаков.
Различают также гипогонадотропный, гипергонадотропный и нормогонадотропный гипогонадизм. Гипергонадотропный гипогонадизм проявляется первичным поражением тестикулярной ткани яичек в сочетании с повышенным уровнем гонадотропных гормонов гипофиза. Гипогонадотропный и нормогонадотропный гипогонадизм возникают при поражении гипоталамо-гипофизарной системы. Гипогонадотропный гипогонадизм связан со снижением секреции гонадотропинов, в результате чего уменьшается выработка андрогенов тестикулярной тканью яичек. Нормогонадотропный гипогонадизм вызван гиперпролактинемией, проявляется нормальным уровнем гонадотропинов и сниженной тестикулярной функцией яичек.
Как первичный, так и вторичный гипогонадизм могут быть врожденными и приобретенными. Проявлением гипогонадизма могут служить некоторые формы мужского бесплодия (от 40 до 60% всех случаев мужского бесплодия). В зависимости от возраста развития недостаточности половых гормонов различаются эмбриональная, допубертатная (от 0 до 12 лет) и постпубертатная формы гипогонадизма.
Врожденный первичный (гипергонадотропный) гипогонадизм встречается:
Приобретенный первичный гипогонадизм развивается в результате воздействия на яички внутренних или внешних факторов после рождения.
Врожденный вторичный (гипогонадотропный) гипогонадизм развивается при состояниях:
Приобретенный вторичный гипогонадизм развивается при:
Причины и механизмы развития гипогонадизма у мужчин
Недостаточность андрогенов может быть вызвана снижением количества вырабатываемых гормонов или нарушением их биосинтеза в результате патологии самих яичек или нарушения гипоталамо-гипофизарной регуляции.
Этиологическими факторами первичного гипогонадизма нередко служат:
Некоторые случаи первичного гипогонадизма являются идиопатическими. Современная эндокринология не располагает достаточными данными об этиологии идиопатического гипогонадизма.
При первичном гипогонадизме происходит снижение уровня андрогенов в крови, развитие компенсаторной реакции надпочечников на гипоандрогенизацию, возрастание продукции гонадотропинов.
К вторичному гипогонадизму приводят нарушения гипоталамо-гипофизарной регуляции (воспалительные процессы, опухоли, сосудистые нарушения, патология эмбрионального развития). Развитие гипогонадизма могут вызывать аденомы гипофиза, продуцирующие гормон роста (при акромегалии) или адренокортикотропный гормон (при болезни Кушинга), пролактинома, послеоперационная или посттравматическая гипоталамо-гипофизарная дисфункция, гемохроматоз, процессы старения, сопровождающиеся возрастным снижением уровня тестостерона крови.
При вторичном гипогонадизме отмечается низкий уровень гонадотропинов, приводящий к уменьшению секреции андрогенов яичками.
Одной из форм мужского гипогонадизма является снижение продукции спермы при нормальном уровне тестостерона, а также крайне редкие случаи снижения уровня тестостерона без уменьшения продукции спермы.
Симптомы гипогонадизма у мужчин
Клинические проявления гипогонадизма обусловлены возрастом возникновения заболевания и степенью андрогенной недостаточности. Нарушение продукции андрогенов во внутриутробном периоде может приводить к развитию двуполых наружных половых органов.
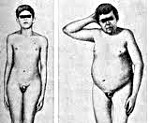
Если поражение яичек произошло у мальчиков в допубертатном периоде, происходит задержка полового развития, формируется типичный евнухоидизм: непропорционально высокий рост, связанный с запаздыванием окостенения эпифизарных (ростовых) зон, неразвитая грудная клетка и плечевой пояс, длинные конечности, слаборазвитая скелетная мускулатура. Может отмечаться развитие ожирения по женскому типу, истинной гинекомастии, гипогенитализма, что проявляется в малом размере полового члена, отсутствии пигментации и складчатости мошонки, гипоплазии яичек, недоразвитии предстательной железы, отсутствии оволосения на лице и лобке, недоразвитии гортани, высоком голосе.
В случаях вторичного гипогонадизма часто возникает ожирение, возможна симптоматика гипофункции коры надпочечников, щитовидной железы, проявления пангипопитуитаризма, отсутствие полового влечения и потенции.
Если снижение функции яичек развивается после завершения полового созревания, то симптомы гипогонадизма выражены слабее. Наблюдается уменьшение размеров яичек, незначительное оволосение лица и тела, жировые отложения по женскому типу, потеря эластичности и истончение кожи, бесплодие, снижение половой функции, вегетативно-сосудистые нарушения.
Уменьшение яичек наблюдается почти во всех случаях мужского гипогонадизма (исключение – если заболевание началось недавно). Уменьшение размеров яичек обычно тесно связано с уменьшением выработки спермы. При утрате спермопродуцирующей функции яичек развивается бесплодие с прекращением продукции тестостерона, снижается либидо, возникает регрессия вторичных половых признаков, эректильная дисфункция, отмечаются генерализованные симптомы (снижение мышечной силы, утомляемость, общая слабость).
Диагностика гипогонадизма у мужчин
Основывается на жалобах пациента, данных анамнеза, исследовании общего статуса с использованием антропометрии, осмотре и пальпации гениталий, оценке клинических симптомов гипогонадизма, степени полового созревания.
По данным рентгенологического исследования оценивается костный возраст. Для определения минеральной насыщенности костей проводится денситометрия. При рентгенографии турецкого седла определяются его размеры и наличие опухоли. Оценка костного возраста позволяет по срокам окостенения лучезапястного сустава и кисти достаточно точно определить начало полового созревания. Начало полового созревания связывают с формированием сесамовидной кости в I пястно-фаланговом суставе (примерно в 13,5 — 14 лет). О полной половой зрелости свидетельствует появление анатомических синостозов. Этот признак дает возможность разграничить допубертатный возраст от пубертатного. Оценивая костный возраст, нужно учитывать возможность более раннего (для пациентов из южных районов) и позднего (для пациентов из северных районов) окостенения, а также то, что нарушение остеогенеза может быть обусловлено и другими факторами. При допубертатном гипогонадизме отмечается отставание на несколько лет «костного» возраста от паспортного.
Лабораторное исследование анализа спермы (спермограмма) при гипогонадизме характеризуется азо- или олигоспермией; иногда эякулят получить не удается. Проводится измерение уровня половых и гонадотропинов: сывороточного тестостерона (общего и свободного), лютеинизирующего, фолликулостимулирующего гормона и гонадолиберина, а также антимюллеровского гормона сыворотки крови, пролактина, эстрадиола. Содержание тестостерона в крови снижено.
Лечение гипогонадизма у мужчин
Лечение гипогонадизма зависит от его клинической формы, выраженности нарушений в гипоталамо-гипофизарной и половой системах, сопутствующих патологий, времени возникновения болезни и возраста постановки диагноза. Терапию гипогонадизма начинают с лечения основного заболевания. Лечение взрослых пациентов состоит в коррекции недостаточности андрогенов и половой дисфункции. Бесплодие, возникшее на фоне врожденного и допубертатного гипогонадизма неизлечимо, особенно в случае аспермии.
Гипогонадизм у женщин
Дефицит эстрогенов вызывает недоразвитие и атрофические изменения женских половых органов, молочных желез, первичную аменорею. Если нарушение в яичниках возникло в допубертатный период, то вторичные половые признаки отсутствуют.
Причинами первичного гипергонадотропного гипогонадизма являются врожденное генетическое нарушение (синдром Шерешевского-Тернера), врожденная гипоплазия яичников, инфекционные процессы (сифилис, туберкулез, эпидемический паротит), ионизирующее излучение (лучевое, рентгеновское), оперативное удаление яичников, аутоиммунное поражение яичников (аутоиммунный оофорит), синдром тестикулярной феминизации (врожденное состояние, при котором внешний вид человека соответствует женщине при мужском генотипе), синдром поликистозных яичников.
Вторичный женский гипогонадизм (гипогонадотропный) возникает при гипоталамо-гипофизарной патологии, характеризуется дефицитом или полным прекращением синтеза и секреции гонадотропинов, регулирующих функцию яичников. Развивается вследствие воспалительных процессов в головном мозге (энцефалит, менингит, арахноидит), повреждающего действия опухолей головного мозга и сопровождается снижением стимулирующего действия гонадотропинов на функцию яичников.
Симптомы гипогонадизма у женщин
Один из основных симптомов гипогонадизма в детородном периоде – нарушение менструального цикла и аменорея. Недостаток женских половых гормонов ведет к недоразвитию половых признаков: гениталий, молочных желез, нарушению отложения жировой клетчатки по женскому типу, скудному оволосению. Если заболевание врожденное, или оно возникло в раннем детском возрасте, то вторичные половые признаки отсутствуют. Характерны узкий таз и плоские ягодицы. Если гипогонадизм развился в пуберантном периоде, половые признаки, которые уже успели развиться, сохраняются, но менструации прекращаются, ткани женских гениталий подвергаются атрофии.
Диагностика гипогонадизма у женщин
При гипогонадизме наблюдается заметное снижение содержания эстрогенов в крови, повышение уровня гонадотропинов (фолликулостимулирующего и лютеинизирующего гормонов). Ультразвуковое исследование выявляет матку, уменьшенную в размерах (гипоплазия матки), уменьшенные яичники. Рентгенография обнаруживает остеопороз или задержку формирования скелета.
Лечение гипогонадизма у женщин
Прогноз для жизни при гипогонадизме – благоприятный. Профилактика гипогонадизма состоит в медико-санитарном просвещении населения, наблюдении беременных женщин и охране их здоровья.
Гипогонадотропный гипогонадизм у женщин: причины возникновения, подходы к диагностике и лечению
ФГБУ Эндокринологический научный центр Минздравсоцразвития России, Москва, Россия; ГБУЗ МО Московский Областной научно-исследовательский клинический институт им. М.Ф. Владимирского, Москва, Россия
Формирующийся в результате нарушения синтеза и/или секреции гонадотропинов гипогонадотропный гипогонадизм может быть органическим (т.е. формирующимся в результате анатомо-функциональных расстройств гипоталамо-гипофизарной области при различных новообразованиях хиазмально-селлярной локализации) или функциональным, приобретенным или врожденным (мутации гена рецептора ГнРГ, β-субъединицы ФСГ; генов SF-1, DAX-1, нарушения в генах рецепторов ЛГ и ФСГ и пр.). В формировании функциональных нарушений принимают участие нейропептид Y (NPY), кортикотропин-рилизинг-гормон (КРГ), лептин, грелин и β-эндорфин. Диагностика заболевания включает определение гонадотропинов, исключение гиперпролактинемии и других эндокринных расстройств, ультразвуковое исследование матки и яичников, магнитно-резонансную томографию головного мозга. При длительности аменореи более года показано исследование плотности костной ткани. При выявлении причин гипогонадотропного гипогонадизма в первую очередь необходимо их устранение (например, ликвидация стресса, исключение физических нагрузок, восстановление нормальной массы тела, или лечение опухолей гипофиза и т.д.), что может приводить к восстановлению менструальной функции и фертильности. Однако в большинстве случаев при стойкой гипоэстрогении у пациенток с гипогонадотропным гипогонадизмом показана эстроген-гестагенная терапия в циклическом режиме, для длительного применения эффективной и безопасной является комбинация 17β-эстрадиола 2 мг и дидрогестерона 10 мг.
«Гипогонадотропным» (или «центральным», «вторичным») называют гипогонадизм, формирующийся в результате нарушения синтеза и/или секреции гонадотропинов. Гипогонадотропный гипогонадизм может быть приобретенным или врожденным заболеванием, органическим или функциональным. Приобретенные органические формы гипогона-дотропного гипогонадизма развиваются в результате анатомо-функциональных расстройств гипоталамо-гипофизарной области при различных новообразованиях хиазмально-селлярной локализации (опухолях гипофиза, краниофарингиомах, и пр.) или других повреждениях (например, при формировании «пустого» турецкого седла) [1]. При этом могут возникать как органические нарушения нейронных путей гипоталамуса, так и изменения непосредственно в клетках аденогипофиза, что, в конечном счете, проявляется развитием функциональной несостоятельности гонадотрофов с нарушением секреции гонадотропинов. Снижение синтеза и секреции гонадотропинов может наблюдаться при геморрагических и ишемических инфарктах гипофиза, гранулематозных и инфильтративных заболеваниях с поражением гипоталамо-гипофизарной области (саркаидозе, туберкулезе, гистиоцитозе Х и др.). Кроме того, возможно развитие аутоиммунного поражения ткани гипофиза – так называемый лимфоцитарный гипофизит [2], при котором происходит диффузная инфильтрация ткани гипофиза лимфоцитами и лимфоидными элементами с развитием деструкции нормальных гипофизарных клеток. Этот процесс может сопровождаться недостаточностью тропных гормонов, в том числе и гонадотропинов. Центральный гипогонадизм может также являться следствием черепно-мозговой травмы (развивается примерно у 25% лиц, перенесших тяжелую или средне-тяжелую травму головного мозга) [3].
Функциональная гонадотропная недостаточность (или так называемая «функциональная гипоталамическая аменорея») является причиной примерно 30–35% случаев вторичной аменореи. В этих случаях причиной гипогонадизма становится снижение частоты и/или амплитуды импульсной секреции гонадотропин-рилизинг-гормона (ГнРГ), возникающее в отсутствие органического поражения гипоталамо-гипофизарной области [4]. Диагностическими критериями функциональной гонадотропной недостаточности можно считать отсутствие менструаций более 6 мес на фоне низких (
Одной из самых распространенных причин функциональной гонадотропной недостаточности является хронический стресс («психогенная» или «стрессорная» аменорея). Другими причинами также считаются чрезмерные физические нагрузки (профессиональный спорт) и критическое снижение массы тела, крайняя форма которого наблюдается при нервной анорексии [4, 5].
В формировании функциональной гипоталамической аменореи принимают участие различные нейропептиды: нейропептид Y (NPY), кортикотропин-рилизинг-гормон (КРГ), лептин, грелин и β-эндорфин, влияющие на секрецию ГнРГ. Патогенез функциональной аменореи при стрессе, избыточных физических нагрузках и потере массы тела сходен, хотя при различных заболеваниях являются ведущими те или иные нарушения секреции нейропептидов. Например, при физических нагрузках и потере массы тела в бóльшей степени наблюдается нарушение секреции NPY, при стресс-индуцированной аменорее и на фоне физических нагрузок – чрезмерная активация КРГ и т.д.
В патофизиологии стрессовой аменореи важную роль играет КРГ. У женщин с функциональной аменореей отмечается повышение секреции КРГ по сравнению со здоровыми женщинами [6], что, с одной стороны, считается косвенным подтверждением хронического стресса, а с другой – фактором нарушения секреции ГнРГ/гонадотропинов. Повышенная секреция КРГ прямо подавляет электрофизиологическую активность импульсного генератора ГнРГ [7]. Кроме того, повышение продукции КРГ стимулирует выработку адренокортикотропного гормона (АКТГ) и β-эндорфина, облада.
Этиопатогенетические аспекты центрального (гипогонадотропного) женского гипогонадизма
Полный текст:
Аннотация
Центральный гипогонадизм (ЦГ) – редкое заболевание, причиной которого является нарушение продукции, секреции или биологического действия гонадотропин-рилизинг гормона (ГнРГ), являющегося главным гормональным регулятором гипоталамо-гипофизарно-гонадной оси у человека. ЦГ у женщин является важной медицинской и социальной проблемой ввиду большого количества бесплодных браков. Этиология данного состояния гетерогенна, и является различной для врожденных и приобретенных форм гипогонадизма. Врожденные формы гипогонадизма имеют генетические причины; на сегодняшний день известно около 50 генов, мутации в которых ассоциированы с ЦГ. В то же время, всего в половине случаев ЦГ удается выявить генетическую основу. Что касается приобретенных форм, их причины разнятся в зависимости от состояния хиазмально-селлярной области (ХСО): при интактном состоянии ХСО можно диагностировать функциональную форму ЦГ, наличие структурных нарушений в этой области говорит в пользу органической причины ЦГ. В данном обзоре изложены современные представления об этиологии и патогенезе центрального гипогонадизма у женщин.
Ключевые слова
Для цитирования:
Локтионова А.С., Иловайская И.А. Этиопатогенетические аспекты центрального (гипогонадотропного) женского гипогонадизма. Медицинский вестник Юга России. 2019;10(4):15-27. https://doi.org/10.21886/2219-8075-2019-10-4-15-27
For citation:
Loktionova A.S., Ilovayskaya I.A. Etiopathogenetic aspects of central (hypogonadotropic) hypogonadism in female. Medical Herald of the South of Russia. 2019;10(4):15-27. (In Russ.) https://doi.org/10.21886/2219-8075-2019-10-4-15-27
Введение
Целью данного обзора является освещение этиологии и патогенеза центрального гипогонадизма у женщин.
Определение и классификация
ЦГ у женщин — это синдром, основным клиническим проявлением которого является аменорея на фоне ги- поэстрогенемии вследствие снижения адекватной (центральной, или гипоталамо-гипофизарной) регуляции работы яичников, при исключении других причин аменореи. Иными словами, при этом состоянии отсутствует нормальная реакция гипофиза в виде повышения уровней гонадотропинов в ответ на снижение концентраций эстрадиола в сыворотке крови. Аменорея при этом может быть как первичной, так и вторичной [8].
Существует два основных этиологических варианта ЦГ: генетически обусловленный и возникший вследствие структурного поражения хиазмально-селлярной области (ХСО). Характеристики указанных вариантов ЦГ представлены в табл. 1.
Этиологическая структура форм центрального гипогонадизма у женщин
Etiology of female central hypogonadism
Генетически обусловленные Genetically determined
Органически обусловленные Structurally determined
Врожденные (СК, нЦГ) Inherited (KS, nCH)
Приобретенные (ФГА) Acquired (FHA)
Манифест до пубертата Start before puberty
Манифест после пубертата Start after puberty
Спонтанное менархе Spontaneous menarche
Присутствует, в старшем возрасте по сравнению со здоровыми добровольцами [8]
Present at an older age compared to healthy women [8]
Картина ХСО по данным визуализирующих методов исследования CSR in radiologic investigation methods
Интактное состояние ХСО Intact CSR
Интактное состояние ХСО Intact CSR
Органическое поражение ХСО Organic damage of CSR
Органическое поражение ХСО Organic damage of CSR
Примечание: СК — синдром Каллманна, нЦГ — нормосмическая форма центрального гипогонадизма, ФГА — функциональная гипоталамическая аменорея, ХСО — хиазмально-селлярная область.
Note: KS — Kallmann syndrome, nCH — normosnic central hypogonadism, FHA — functional hypothalamic amenorrhea, CSR — chiasmosellar region.
Врожденные формы центрального гипогонадизма
С момента обнаружения в 1991 г. первой генной аберрации, ассоциированной с СК (мутация гена KAL1 (ANOS)) [19], ЦГ считался заболеванием с моногенным типом наследования. Но данные последних двух десятилетий, отмеченных повышенным интересом к поиску причин ЦГ, привели к тому, что представление о моно- генном наследовании меняется в пользу так называемого «олигогенного», когда у одного пациента можно выявить мутации сразу нескольких ответственных за развитие ЦГ генов [20]; олигогенная этиология выявляется примерно в каждом пятом случае врожденного ЦГ [16]. При этом гены проявляют некоторую специфичность: в то время как альтерации в одних приводят к СК, мутации в других фенотипически выражаются как нЦГ, и есть группа генов, мутации в которых обнаруживают и при СК, и при нЦГ [13]. Учитывая особенности онтогенеза ГнРГ-нейронов, некоторые исследователи предлагают дифференцировать так называемые «нейроразвивающие» гены, отвечающие за нормальные развитие и миграцию этих нейронов, и «нейроэндокринные», влияющие на выработку ГнРГ и оказание им биологического эффекта на гонадотрофы гипофиза. Очевидно, что нейроразвивающие гены большей частью ответственны за развитие фенотипа СК.
Нейроразвивающие гены
Итак, слаженную работу репродуктивной оси координирует ГнРГ, который вырабатывается нейронами ар- куатного ядра гипоталамуса. Всего в гипоталамусе примерно 1500 ГнРГ-нейронов [13]. Во время эмбриогенеза эти нейроны зарождаются в медиальной обонятельной плакоде, затем мигрируют сквозь пластинку решетчатой кости, через развивающиеся обонятельные луковицы, и затем, после прохождения через полушария переднего мозга, заканчивают путь в гипоталамусе, где в течение жизни и выполняют свою функцию [11]. Уже упомянутый KAL1 (ANOS1) как раз играет очень важную роль в этой миграции: продукт этого гена, белок аносмин, ассоциирован с нейрональной клеточной адгезией и аксональной миграцией. Результатом мутаций в этом гене является дисфункция аносмина, вследствие которой нарушается процесс миграции ГнРГ-нейронов в гипоталамус. Аносмин экспрессируется не только в центральной нервной системе, но также и в пищеварительной, дыхательной, мочеполовой, сердечно-сосудистой и опорнодвигательной системах [21]. Именно этой широтой экспрессии гена KAL1 в различных органах и тканях можно объяснить наличие внерепродуктивных фенотипических признаков СК, таких как агенезия почек или синкинезии (патологические зеркальные движения конечностей).
Примерно через 10 лет после открытия гена KAL1 количество генов-кандидатов на роль причины ЦГ стало постепенно расти. При изучении генотипов пациентов, страдающих недостаточностью ГнРГ, выявлены новые нейроразвивающие гены. Одним из первых был ген рецептора 1 фактора роста фибробластов, FGFR1. После открытия роли этого гена в патогенезе ЦГ, было описано множество мутаций его экзонов [22]. Несмотря на наличие 23 известных лигандов этого рецептора (белков ФРФ, факторов роста фибробластов), только ФРФ-8 был описан как лиганд, ответственный за нейрональную миграцию; мутации гена FGF8 также были найдены у пациентов с ЦГ [23]. Таким образом, продукты генов FGF8 и FGFR1 также имеют важное значение для нормальной миграции ГнРГ-нейронов. Чуть позже, при изучении бе- лок-белковых взаимодействий, был выявлен еще ряд генов, продукты которых необходимы для развития ГнРГ- нейронов: FGF17, IL17RD, DUSP6, SPRY4, GLCE, и FLRT3, а также лиганды рецептора IL17RD [24].
Еще одна важная пара лиганд-рецептор — это PROK2 и PROKR2. Ген PROKR2 (он же KAL3) кодирует рецептор, связанный с G-белком, рецептор-2 прокинетицина; ген PROK2 (он же KAL4) кодирует белок прокинетицин 2 (РК2). При взаимодействии этой пары запускается сигнальный киназный каскад, необходимый для нейрогенеза и миграции ольфакторных и ГнРГ-нейронов [25]. Также работа этой лиганд-рецепторной пары участвует в высвобождении ГнРГ нейронами гипоталамуса.
В сумме, порядка 30 генов, известных на данный момент в качестве причины ЦГ, можно отнести к нейроразвивающим. Среди них, кроме уже перечисленных, гены, участвующие в процессах межклеточного взаимодействия, ответственные за аксональный рост и за миграцию клеток нервного гребня, а также различные транскрипционные факторы [13].
Нейроэндокринные гены
Ген самого ГнРГ (GNRH1) и рецептора к нему (GNRHR) являются, пожалуй, самыми очевидными кандидатами на роль причины ЦГ. Действительно, у ряда пациентов с врожденным ЦГ были выявлены мутации в гене GNRHR с различными паттернами наследования — аутосомно- рецессивным моногенным и олигогенным [26]. Вариации GNRH1, также приводящие к фенотипу нЦГ, встречаются намного реже; впервые мутации этого гена были обнаружены в крупном исследовании с участием более чем 300 пациентов с нормосмической формой врожденного гипогонадотропного гипогонадизма [27].
В 2003 г. две независимые группы ученых одновременно выявили аутосомно-рецессивный тип наследования мутации гена KISS1R (ранее известного как GPR54), которые приводят к развитию нЦГ [28,29]. Белок KISS1R — рецептор, основным лигандом которого является кис- спептин-1 (ранее также известный как кисспептин-54). Ген кисспептина-1 (KISS1) — также является исключительно важным в работе репродуктивной оси, и мутации в нем описаны как одна из причин нЦГ [30]. Белок кис- спептин был открыт в 1996 г. как супрессор метастазирования меланомы: его мРНК была выделена из тканей неметастазирующей меланомы, в то время как в тканях метастазирующей опухоли её выявлено не было. В связи с этим белок получил свое первое название «метастатин» [31]. В последующем было показано, что кисспептин се- кретируется KISS-нейронами гипоталамуса, и было изучено его прямое и опосредованное стимулирующее влияние на секрецию ГнРГ. На данный момент эта лиганд- рецепторная пара является, без преувеличения, самым мощным известным регулятором работы ГнРГ-нейронов. В ходе научных работ эта исключительная роль кисспеп- тина в репродуктивной функции млекопитающих была подтверждена и у животных [32], и у человека: в исследовании на контрольной группе здоровых добровольцев-мужчин было продемонстрировано дозо-зависимое увеличение уровня ЛГ при введении им кисспептина-10, и, в меньшей степени, прирост уровней ФСГ и тестостерона [33]. Измерение ГнРГ в крови напрямую является технически сложным ввиду его малого содержания в периферической крови, поэтому влияние кисспептина на выработку ГнРГ оценивалось опосредованно через повышение уровней гонадотропинов. Помимо всего прочего, увеличение импульсной секреции кисспептина инициирует начало полового созревания у человека [34].
Заметный прогресс в области идентификации генов, сопряженных с различными заболеваниями, был достигнут благодаря широким исследованиям GWAS (genome-wide association study, полногеномный поиск ассоциаций). Например, при проведении подобного исследования по определению генетических предикторов возраста наступления менархе и менопаузы, были выявлены гены, локусы которых находятся в непосредственной близости от локусов генов, сопряженных с ЦГ [38][39]. С учетом того, что на данный момент лишь не более чем в половине случаев ЦГ удается выяснить генетическую причину, в ближайшее время стоит ожидать появления новых генов-кандидатов на роль причины ЦГ.
Известные на сегодняшний день гены, мутации которых выявлены у пациентов с ЦГ, перечислены в табл. 2.
Гены, ответственные за развитие ЦГ и их характеристики ([13], адаптировано)
Genes responsible for CH development and their characteristics ([13], adapt.)