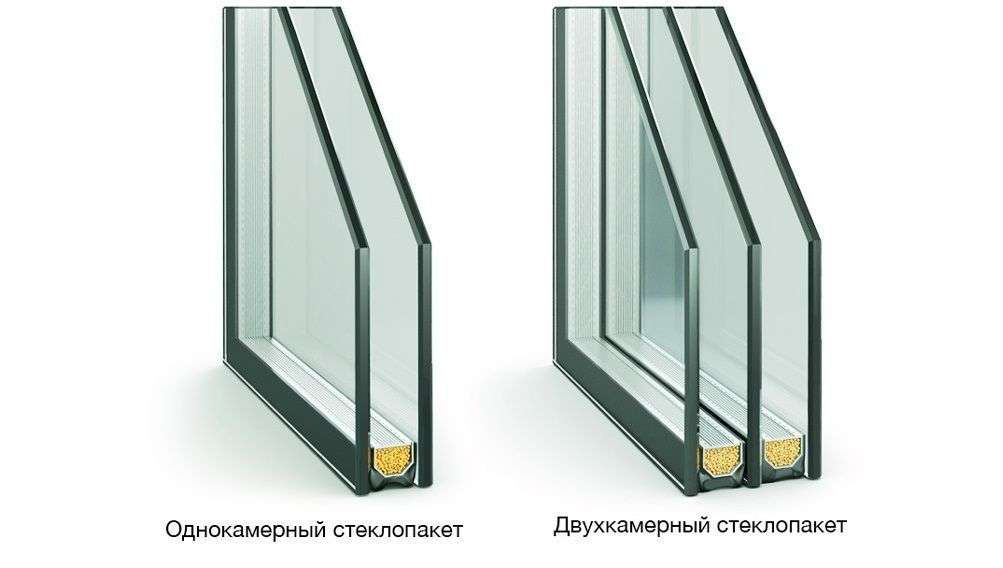
Нейрональный цероидный липофусциноз
Нейрональный цероидный липофусциноз (НЦЛ) – это группа генетических заболеваний, в основе которых лежит накопление в клеточных структурах нейронов и других тканей токсического пигмента – липофусцина. Патология наследуется по аутосомно-рецессивному типу.
Патогенез нейронального цероидного липофусциноза
В основе патогенеза лежит нарушение утилизации пигмента липофусцина. Он накапливается в тканях организма человека и в норме, но гораздо медленнее. В случае нейронального цероидного липофусциноза это накопление происходит стремительно и приводит к атрофии тканей. Липопигменты локализуются в клеточных органеллах – лизосомах, выполняющих функцию утилизации отработанных клеточных элементов.
Ранее в основе классификации нейронального цероидного липофусциноза лежал возраст пациента, в котором начинало манифестировать заболевание. В зависимости от этого выделяли 4 типа патологии:
По мере изучения наследственных болезней были выявлены мутации в генах NCL, которые и легли в основу современной классификации. В настоящее время выделяют 10 типов заболевания в зависимости от имеющихся генетических нарушений.
Клинические проявления нейронального цероидного липофусциноза
Нейрональный цероидный липофусциноз является прогрессирующим заболеванием детей и подростков, однако некоторые формы патологии могут развиваться и у взрослых людей младше 40 лет. До появления клинических проявлений дети ничем не отличаются от своих сверстников.
Манифестация заболевания проявляется одним из трех признаков:
По мере прогрессирования присоединяются следующие признаки:
Со времени возникновения первых симптомов состояние ребенка быстро ухудшается. Гибель происходит в течение нескольких лет. При благоприятных формах нейронального цероидного липофусциноза пациенты редко переживают сорокалетний рубеж.
Методы лечения нейронального цероидного липофусциноза
На сегодняшний день эффективного патогенетического лечения нет, однако состояние пациентов можно облегчить с помощью симптоматической терапии. Это назначение противоэпилептических препаратов, миорелаксантов, седативных, антипсихотических, обезболивающих средств, витаминов. Иногда временное улучшение приносит физиотерапия.
В некоторых странах ведется активный поиск методов лечения, который сосредоточен на трех направлениях:
В целом есть обнадеживающие результаты, позволяющие надеяться на победу страшной болезни в будущем. В США и Европе зарегистрирована фермент-заместительная терапия рекомбинантной альфа-церлипеназой (Бринейра). Препарат позволяет прекратить прогрессирование заболевания.
В рамках диагностического поиска используется нейровизуализация (МРТ, ПЭТ), ЭЭГ, электроретинограмма. Активно применяется гистологическое исследование, но наиболее ценной информацией обладает молекулярно-генетическая диагностика для определения мутаций в генах. Исследуется материал от обоих родителей и ребенка. На основании полученных данных делается заключение о наличии или отсутствии у пациента нейронального цероидного липофусциноза.
Нцл 2 типа что это
Это заболевание в основном затрагивает детей и подростков, однако есть редкие формы НЦЛ, проявляющиеся у взрослых.
В зависимости от типа или формы НЦЛ, это заболевание может проявляться в различное время развития ребенка или подростка. Так НЦЛ 1 дает о себе знать к концу первого года жизни малыша, НЦЛ 2 –примерно в 2-4 года, НЦЛ3- 8-10лет.
Как правило, такие дети рождаются здоровыми и в первые годы своей жизни, до начала заболевания, достигают многих успехов в развитии. Они хорошо развиты физически, контактны, посещают детские учреждения, психологически развиты по возрасту. Однако может наблюдаться небольшое отставание в развитии речи или некоторых навыков.
НЦЛ может проявляться постепенно, начало заболевания определяется одним из трех признаков: судороги, атаксия (неуклюжесть походки) и снижение зрения. Эти симптомы могут быть при очень многих других заболеваниях, а болезнь Баттена настолько редко встречается, что почти всегда её диагностируют не сразу. То есть сначала ребенку ставят неправильный основной диагноз, как правило, эпилепсию или какое-либо заболевание глаз.
Тем не менее, по мере прогрессирования заболевания вскоре становится очевидно, что первоначальный диагноз неверен и начинается более серьезное обследование больного ребенка.
Прогрессирование заболевания заключается в ухудшении зрения, потери навыков, речи, потери способности стоять и ходить самостоятельно. Как правило, сначала ребенку делают МРТ головного мозга и по данным такого исследования опытный врач-невролог может предположить наличие заболевания. Если данные МРТ (атрофия больших полушарий и мозжечка) позволяют сделать выводы о наличии заболевания, ребенка и его родителей отправляют на генетическую экспертизу.
НЦЛ является генетическим заболеванием. Различные формы его вызваны дефектами (мутациями) в различных генах NCL. НЦЛ-генетическое прогрессирующее заболевание головного мозга детей и подростков. NCL заболевания, которые встречаются в зрелом возрасте, очень редки и четко не определены.
В случае НЦЛ 2 типа из-за наличия мутантного гена NCL2 снижено содержание фермента TPP1 (трипептидил пептидазы 1) или же фермент отсутствует вообще (TPP1-лизосомный фермент). Как следствие, не разрушается липофусцин и образуется цероид в мозге. Следует отметить, что эти включения могут накапливаться не только в нейронах, но и в лимфоцитах, биоптатах кожи, конъюктивы, слизистой прямой кишки и клетках сетчатки, сердечной мышце, печени. Однако, при НЦЛ это происходит только в нейронах и почему- до сих пор неизвестно… Под действием липофусцина погибают нейроны, что приводит к атрофии больших полушарий мозга и мозжечка. Это хорошо видно на снимках МРТ. И именно данные МРТ наряду с клиническими проявлениями позволяют врачам предположить наличие болезни.
АУТОСОМНО-РЕЦЕССИВНЫЙ ТИП НАСЛЕДОВАНИЯ
Наследственные заболевания- это болезни, обусловленные нарушениями в процессах хранения, передачи и реализации генетической информации. С развитием генетики человека, в том числе и генетики медицинской, выяснилась наследственная природа многих заболеваний и синдромов, считавшихся ранее болезнями с неустановленной этиологией. Роль наследственных факторов подтверждается более высокой частотой ряда заболеваний в некоторых семьях по сравнению с населением в целом.
В основе наследственных заболеваний лежат мутации — преимущественно хромосомные и генные, соответственно чему условно говорят о хромосомных болезнях и собственно наследственных (генных) болезнях. Мутация ведёт к нарушению синтеза определенного полипептида (структурного белка или фермента). В зависимости от того, какова роль этого полипептида в жизнедеятельности организма, у больного возникают нарушения (изменения фенотипа) локального или системного порядка.
Наследственно дегенеративные болезни отличаются деструктивными и дистрофическими изменениями в тканях (нервной, мышечной и др.), избирательностью поражения нервной системы, мышц, внутренних органов и кожи, прогрессирующим течением.
Одни из них возникают с первых лет жизни, другие — много лет спустя после рождения.
Типы наследования различны: аутосомно-доминантный, аутосомно-рецессивный и рецессивный, сцепленный с полом. В зависимости от того, где локализован патологический (мутантный) ген — в аутосоме или в половой хромосоме — и каковы его взаимоотношения с нормальным аллелем, т. е. является ли мутация доминантной (нормальный ген подавляется патологическим) или рецессивной (патологический ген подавляется нормальным) и различают типы наследования.
При заболеваниях, наследуемых по аутосомно-рецессивному типу, мутантный ген проявляется лишь в гомозиготном состоянии; больные мальчики и девочки рождаются с одинаковой частотой; родители больных фенотипически здоровы, но являются гетерозиготными носителями мутантного гена; патологическая наследственность прослеживается в родословной семьи «по горизонтали»; вероятность рождения больных детей возрастает в случае кровного родства родителей. Если один из родителей гомозиготен по патологическому рецессивному гену, а другой является его гетерозиготным носителем, то в половине случаев дети могут оказаться больными, и создаётся впечатление наследования заболевания по доминантному типу. Такое явление носит название псевдодоминирования. От истинного доминирования оно отличается тем, что больные с рецессивной мутацией в браке со здоровыми людьми всегда будут давать здоровое потомство, а здоровые в браке с гетерозиготными носителями с определенной частотой (25%) будут иметь больных детей.
IPMN-карцинома
 Кто входит в группу риска?
Кто входит в группу риска?
Основной фактор риска – наличие кистозной опухоли поджелудочной железы (IPMN). Малигнизация чаще возникает в пожилом возрасте, у пациентов старше 65 лет. Мужчины болеют чаще. Помимо этого, курение, прием алкоголя, наличие хронического панкреатита являются факторами риска развития болезни.
Заподозрить заболевание можно при возникновении следующих симптомов:
При появлении любого из этих признаков, необходимо обратиться к врачу. Нельзя заниматься каким-либо самолечением, так как указанные симптомы могут быть следствием большого числа других заболеваний. Специалист оценит ситуацию и назначит нужное обследование.
Для диагностики IPMN-карциномы применяется следующие методы.
Прежде всего, так называемые неинвазивные методы, когда обследование проводится без того, или иного проникновения в организм и практически отсутствуют осложнения
К инвазивным (с проникновением в организм) методам относится эндосонография (УЗИ через эндоскоп) с возможной биопсией.
Среди лабораторных показателей, помимо биохимических, важны данные опухолевых маркеров СЕА и СА19-9. Они имеют не только диагностическое, но и прогностическое значение.
Данные обследования в сочетании с клиническими данными и учетом общего состояния больного позволяют определить рациональную тактику лечения.
В Московском клиническом научно-практическом центре имени А.С.Логинова этот вопрос решается в каждом конкретном случае на консилиуме (совещании) всех необходимых специалистов, прежде всего онкологов и хирургов.
В ряде случаев на первом этапе показано хирургическое лечение с/без последующей химиотерапией. В нашем центре выполняют современные лапароскопические операции, в том числе и с применением робота DaVinci. Накоплен самый большой в России и один из самых больших в мире опыт лапароскопических операций на поджелудочной железе.
В других случаях лечение начинается с химиотерапии, а затем в зависимости от её результатов вновь обсуждается вопрос об операции. В настоящее время в Центре трудится команда ведущих онкологов России, располагающих необходимыми современными средствами лечения.
Не откладывайте свой визит к специалистам нашего Центра. Это поможет своевременно диагностировать заболевание и правильно выбрать тактику лечения. Врачи отделения высокотехнологичной хирургии и хирургической эндоскопии и отделения химиотерапии ГБУЗ МКНЦ имени А.С. Логинова ДЗМ всегда готовы Вам помочь.
Антинуклеарный фактор на клеточной линии HEp-2 (АНФ)
Описание
Антинуклеарный фактор на клеточной линии HEp-2 (АНФ) — это тест для выявления антинуклеарных антител методом непрямой иммунофлюоресценции.
При системной красной волчанке (СКВ) и других системных ревматических заболеваниях, иммунный ответ направлен против нуклеопротеиновых антигенов, т.е. комплексов нуклеиновых кислот и белков. Эти эндогенные нуклеопротеиновые комплексы образуются в ходе процесса апоптоза эпителиоцитов и напоминают чужеродные вирусные частицы. Ускорение процессов апоптоза под действием ультрафиолетового облучения, вирусных инфекций и лекарственных препаратов, запускает аутоиммунные ответы при СКВ. При апоптозе основные антигены антител конденсируются в апоптотических тельцах, которые становятся мишенью для аутоантител. В настоящее время описаны около 200 разновидностей антител к нуклеопротеинам и рибонуклеиновым кислотам, которые получили название антинуклеарные антитела. При аутоиммунных заболеваниях аутоантитела к ядерным антигенам не обладают прямым цитотоксическим действием на клетки человека, однако иммунные комплексы способны запускать иммунологическое воспаление особенно в местах, где сосуды особенно тонки, в том числе в почках, коже, ЦНС, синовиальной оболочке суставов, плевре.
Исследование антинуклеарного фактора (АНФ) представляет собой основной метод выявления антинуклеарных антител, позволяя выявлять аутоантитела к нуклеиновым кислотам (дсДНК, осДНК, РНК), рибонуклеопротеинам, а также большинству конформационных и нерастворимых антигенов.
Антинуклеарные антитела обнаруживаются благодаря их связыванию с внутриклеточными антигенами перевиваемой линии клеток эпителия человека (HEp-2). Ядро и цитоплазма клеток НЕр-2 содержит все антигены, характерные для человеческого организма, что позволяет выявлять в одном тесте все основные антинуклеарные антитела. Метод непрямой иммунофлюоресценции на клеточной линии НЕр2 рекомендован в качестве золотого стандарта выявления антинуклеарных антител ведущими европейскими (EASIgroup2010) и американскими экспертами (ACRANATaskforce2008). В силу многообразия антигенов антинуклеарных антител, не все они могут быть очищены или синтезированы для использования в иммуноферментных тестах по выявлению антинуклеарных антител.
Позитивность по АНФ на клеточной линии НЕр-2 отмечается при системной красной волчанке (СКВ), системных ревматических заболеваниях и многих аутоиммунных заболеваниях, что делает его универсальным тестом в обследовании больных с аутоиммунной патологией. Положительный результат АНФ отмечается более чем у 90% больных с диффузными болезнями соединительной ткани, такими как системная красная волчанка и кожные формы этого заболевания, склеродермия и ее разновидности, смешанное заболевание соединительной ткани, синдром Шегрена. Выявление АНФ имеет большое значение в диагностике ювенильного ревматоидного артрита и аутоиммунных заболеваний печени. Представители этого семейства аутоантител могут быть обнаружены при множестве других аутоиммунных (тиреоидит, диабет), инфекционных (вирусный гепатит), воспалительных и онкологических заболеваний. Встречаемость АНФ достигает 1–3% у клинически здоровых людей и несколько возрастает у лиц старше 65 лет. Лица с высокими титрами АНФ имеют повышенный риск развития аутоиммунных заболеваний.
Интерпретация результатов
Единицы измерения: титр.
Диабет ни 1, ни 2. «Новый диабет». Сахарный диабет полтора (1.5)

В настоящее время наблюдается тенденция к появлению форм сахарного диабета с нетипичным, не классическим течением.
Вопрос этот я поднимаю и пытаюсь вам донести потому, что кроме известных всем сахарного диабета 1 типа, сахарного диабета 2 типа, стали часто встречаться LADA-диабет (сахарный диабет полтора, 1.5) и MODY-диабет, отдельно выделяют диабет БЕРЕМЕННЫХ (гестационный диабет).
Теперь по порядку, для того чтобы разобраться, что такое сахарный диабет полтора ( 1.5), надо уточнить, что такое сахарный диабет 1 и 2 типа.
Сахарный диабет 1 типа – составляет не более 10 % от всего сахарного диабета, известный термин его обозначающий – инсулинозависимый, (начинается чаще с детского возраста, но сейчас встречается и с началом в позднем возрасте), при этом типе диабета на фоне аутоиммунного воспаления поджелудочной железы, почти полностью гибнут в-клетки, вырабатывающие инсулин, человек не может жить без инъекций инсулина, часто «впадает» в кетоацидоз (состояние с образованием ацетона в организме из распада жиров, интоксикацией и обезвоживанием).
Этот тип диабета бывает 2 видов:
Сахарный диабет 2 типа, известен больше других типов сахарного диабета. Этот сахарный диабет составляет 80% от всего диабета. Также бывает 2–х подтипов (с преобладанием инсулинорезистентности у лиц с ожирением и с преобладанием нарушения секреции инсулина, чаще у худых). Аутоантител при данном виде сахарного диабета в крови нет. Кетоацидоза не бывает в начале заболевания, как при СД1, он может развиться через 20-30 лет от начала заболевания, при истощении поджелудочной железы и прекращении выработки инсулина. Лечится СД2, как известно, сочетанием разных групп сахароснижающих препаратов.
При его диагностике сдают кровь на следующие антитела:
Тем самым уточняется сохранность в-клеток поджелудочной железы и секреции инсулина, основного гормона усваивающего сахар пищи). Это поможет поставить диагноз и подобрать верное лечение.