Тубулопатия у детей: причины, симптомы, лечение

Тубулопатия у детей – это целая группа гетерогенных болезней мочевыводящей системы, которые связаны с проблемами реабсорбции в почечных канальцах. Клиническая картина очень разнообразная. Эта патология проявляется симптомами нехватки определенных микроэлементов, то есть у ребенка начинается слабость, обезвоживание, колебание давления, полиурия, гипотония мышц и пр. Также могут возникать симптомы, которые характерны для рахита. Диагностика предполагает не только лабораторные анализы и рентгенологическое исследование, но и изучение здоровья семьи в нескольких поколениях. Лечение является симптоматическим и направлено на то, чтобы устранить дефицит микроэлементов в организме ребенка.
Частота тубулопатии разная, особенно с учетом того, что это не один недуг, а целая группа, к примеру, синдром Барттюра встречается очень редко, а вот почечная глюкозурия обнаруживается у 6% новорожденных малышей. Актуальность этой проблемы является очень высокой. Во-первых, заболевание протекает так, как будто это рахит, а по определенным причинам диагностируют его часто уже на стадии, когда почечная недостаточность становится хронической, то есть ее уже практически невозможно корректировать. Во-вторых, многие разновидности и отдельные синдромы, которые относятся к тубулопатии у детей, так и не изучены полностью, а поэтому доступна исключительно терапия поддерживающего типа.
Причины
Классификация тубулопатии у детей в зависимости от причин, вызывающих этот недуг, может быть врожденной (называется еще первичной) и приобретенной (известна также как вторичная). Первичная развивается из-за следующих факторов:
Вторичная тубулопатия представляет собой следствие таких процессов:
Канальцы передают полезные микроэлементы почкам. Если в них есть какие-либо нарушения, закупорки, то это приводит к тяжелым патологиям. Нельзя игнорировать заболевания, так как последствия плачевные. Возможен даже летальный исход.
Классификация
В первую очередь тубулопатия бывает первичной и вторичной. Такая классификация основана на этиологическом принципе. Первичная является врожденной или наследственной, а вторичная возникает из-за негативного воздействия на почечные канальца извне, то есть патология вызвана болезнями других органов и систем. Это могут быть даже опухоли, ожоговая болезнь, патологии крови и пр.
Часто используется классификация в зависимости от уровня поражения системы канальцев. Выделяют такие разновидности, где в основном повреждены следующие части:
Также существует классификация тубулопатии по патанатопии. Заболевание отличается в зависимости от ведущего симптома болезни, то есть возможна полиурия, нефролитиаз, деформации скелета и прочее.
Симптомы
Тубулопатия у детей и ее клинические проявления напрямую связаны с транспортировкой полезных микроэлементов, а также тем, какая часть общей системы нарушена, к примеру, если у пациента диабет гипофосфатемического типа, то в этом случае он страдает от того, что с мочой в большом количестве выводится кальций.
Также скелетные изменения, характерные для рахита, развиваются еще и при заболевании де Тони-Дебре-Фанкони. Однако в данном случае они не единичны, а появляются совместно с множественными деформациями костей, патологий внутренних органов (сердце, кровеносные сосуды, глаза, органы дыхательной и пищеварительной системы).
Псевдогипоальдостеронизм – это заболевание, для которого характерно чрезмерное выведение хлорида натрия из организма. Вещество покидает организм не только вместе с мочой, но и слюной, каловыми массами. Из-за этого ребенок страдает от сильного обезвоживания.
Еще один синдром – это недостаток глюкозы при тубулопатии. В этом случае ребенок становится слабым, часто чувствует голод. Кроме того, проявляется полидипсия и полиурия. Из-за этого при почечной глюкозурии возникают симптомы, которые похожи на сахарный диабет.
Если снижается впитывание калия, то развивается гипокалиемия. Она тоже относится к тубулопатии. Вследствие нее развивается мышечная гипотония, а в более тяжелых формах – даже паралич.
Если нарушается процесс транспортировки аминокислот через почечные мембраны, то это приводит к их дефициту, который проявляется определенными симптомами, к примеру, цистинурия сопровождается почечными коликами. Синдром Хартнапа характеризуется симптомами мозжечковой атаксии и пеллагры. Часто это сопровождается нефролитиазом.
Когда следует обратиться к врачу
Обратиться к врачу необходимо, если появляются странные симптомы, но при этом у новорожденного не обнаружили других болезней, к примеру, это касается изменений тела, которые характерны для рахита. Также нужно обращать внимание на постоянное чувство голода, слабость, обезвоживание, почечные колики. Еще могут появляться признаки сахарного диабета или проблемы с внутренними органами.
На ранних стадиях все синдромы, которые относятся к тубулопатии, практически не удается диагностировать. Клиническая картина у них крайне неточная и размытая. Болезнь может маскироваться под похожую или сопутствующую патологию. Из-за этого ее чаще всего выявляются уже, когда почечная недостаточность ярко выражена.
При тубулопатии у детей клинические рекомендации и диагностику проводит врач-педиатр. Кроме того, чтобы пройти полное обследование, пациента направляют также на консультацию к другим специалистам с более узким профилем – например, кардиологу, хирургу-травматологу, офтальмологу и пр. На основе всех результатов исследований они ставят точный диагноз и подбирают необходимые меры по урегулированию проблемы.
Диагностику тубулопатии можно провести в АО «Медицина» (клиника академика Ройтберга), которая располагается в центре Москвы.
Лечение
Терапия тубулопатии у детей осуществляется только в стационарных условиях под строгим наблюдением доктора. Так как обычно заболевание вызвано генетическими дефектами, то не разработана этиотропная терапия. Если заболевание носит вторичный характер и развивается на фоне неправильного питания, болезней накопления, то предрасполагающие факторы поддаются корректировке, что позволяет улучшить общее состояние больного. К примеру, если есть ожоги в тяжелой степени, то необходим гемодиализ. Ферментная терапия заместительного типа назначается при лизосомных болезнях накопления. Подобное лечение позволяет предупредить осложнение веществ в почечных канальцах. Дополнительно всегда назначается диета ощелачивающего типа. Рекомендуется употреблять фрукты, молочные продукты, капусту, картофель.
Диагностика
Если есть подозрение на тубулопатию у ребенка, то педиатр подбирает комплекс мер для изучения мочи и работы мочевыводящей системы. В обязательном порядке требуется выявить уровень содержания кальция, калия, натрия, хлора в урине, а также сделать тест на наличие глюкозы в ней. Хромография проводится с целью определения аминокислот. Конкременты изучают под микроскопом либо выявляются во время УЗИ, рентгенографии. Также процедура УЗИ позволяет определить аномалии в развитии мочевыводящих органов.
Также исследуется кровь ребенка. При увеличении выделений веществ в почках в крови наблюдается их дефицит. Для этого проводится биохимический анализ крови. Также изучается уровень глюкозы и гормональных веществ в крови. Что касается последнего, то особое внимание отводится соединениям, которые вырабатываются надпочечниками и щитовидной железой. Именно эти гормоны активно влияют на обменные процессы кальция, калия, натрия и пр. Для определения первичной тубулопатии необходимо провести молекулярно-генетический анализ родословной.
Как записаться к педиатру
Чтобы посетить педиатра, пройти детальное обследование при подозрении на тубулопатию в АО «Медицина» (клиника академика Ройтберга), вы можете позвонить нам по телефону +7(495)995-00-33, заполнить специальную онлайн-заявку на сайте.
Адрес клиники: 2-й Тверской-Ямской пер. 10.
Тубулопатии

Тубулопатии – гетерогенная группа заболеваний мочевыделительной системы, связанная с нарушением процесса реабсорбции в почечных канальцах. Клиника крайне разнообразна. Тубулопатии проявляются признаками дефицита того или иного микроэлемента: слабостью, обезвоживанием, полиурией, мышечной гипотонией, симптомами рахита, изменением артериального давления в ту или иную сторону и т. д. Проводится исследование мочи и функции почек, лабораторная и рентгенологическая диагностика, анализ родословной. Лечение тубулопатий чаще симптоматическое, направлено на нормализацию уровня натрия, калия, кальция и др.
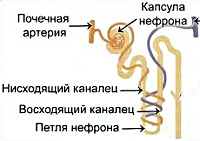
Общие сведения
Несмотря на то, что тубулопатиями считаются заболевания, при которых имеется нарушение работы почечных канальцев, часть нозологий данной группы на самом деле являются энзимопатиями, то есть в их основе лежит нарушение структуры белков-переносчиков. Частота встречаемости тубулопатий сильно варьирует. Например, синдром Барттера диагностируется крайне редко, в то время как частота почечной глюкозурии достигает 6% среди новорожденных (с учетом семейной формы). Актуальность проблемы в педиатрии чрезвычайно высока. Во-первых, тубулопатии часто протекают под маской рахита и по этой причине диагностируются уже на стадии хронической почечной недостаточности, которая практически не поддается коррекции. Во-вторых, причина большинства первичных тубулопатий остается не до конца изученной, а значит, возможна только поддерживающая терапия.
Причины тубулопатий
Большинство тубулопатий возникают вследствие генетического дефекта. Мутация может происходить в генах, кодирующих синтез белков, транспортирующих различные микроэлементы через мембрану клеток почечных канальцев. Также возможно отсутствие реакции на гормоны, регулирующие реабсорбцию калия, натрия, бикарбонатов и других соединений, что связано с дефектом развития рецепторов к этим гормонам на клеточных мембранах. Еще одной причиной тубулопатий являются диспластические изменения в почках, то есть неправильная закладка тканей во внутриутробном периоде, из-за чего изменяется сама структура мембран тубулоцитов.
Вышеперечисленные факторы приводят к нарушению процесса реабсорбции различных соединений и микроэлементов из первичной мочи. Однако то же самое может происходить в почечных канальцах после отравлений солями тяжелых металлов или лекарственными препаратами, при болезнях накопления (сфинголипидозы, мукополисахаридоз и др.), когда также имеет место генетический дефект, который в данном случае находится за пределами почек и мочевыделительной системы. Любые заболевания, способные привести к нарушению функционирования почечных канальцев, могут спровоцировать тубулопатии. Это могут быть ожоги тяжелой степени, болезни крови и другие состояния, при которых нагрузка на почки значительно повышается.
Классификация тубулопатий
Разделение тубулопатий на первичные и вторичные основано на этиологическом принципе. В первом случае речь идет о наследственных и врожденных патологиях в системе почечных канальцев, в то время как вторичные тубулопатии связаны с влиянием извне, то есть с заболеваниями других органов и систем, болезнями крови, опухолями, ожоговой болезнью и т. д. Кроме этого, существует клиническая классификация, основанная на ведущем синдроме заболевания: полиурии, скелетных деформациях, нефролитиазе и других симптомах. На практике часто используется классификация по уровню поражения канальцевого аппарата. Выделяют тубулопатии:
Симптомы тубулопатий
Клинические проявления зависят от того, транспорт какого именно соединения нарушается. Так, при гипофосфатемическом диабете наблюдается избыточное выведение с мочой кальция, поэтому в клинике превалируют симптомы рахита, высокорезистентного к терапии витамином D. Рахитоподобные изменения развиваются также при болезни де Тони-Дебре-Фанкони, но в данном случае они сочетаются с множественными костными деформациями и патологиями со стороны других органов (глаз, сердца и сосудов, ЛОР-органов и др.). Псевдогипоальдостеронизм характеризуется избыточным выведением хлорида натрия из организма, причем не только с мочой, но и с калом, слюной, потом, поэтому у больных наблюдаются симптомы обезвоживания.
Дефицит глюкозы при тубулопатиях проявляется слабостью, чувством голода, а также полиурией и полидипсией, из-за чего почечная глюкозурия сопровождается симптомами, напоминающими сахарный диабет. Снижение реабсорбции калия с последующей гипокалиемией может стать причиной мышечной гипотонии вплоть до развития параличей. Нарушение почечного мембранного транспорта аминокислот провоцирует их дефицит, проявляющийся теми или иными симптомами. Так, при цистинурии наблюдаются почечные колики, при болезни Хартнапа – признаки мозжечковой атаксии и пеллагры. Данная группа тубулопатий часто приводит к нефролитиазу.
Диагностика тубулопатий
При подозрении на тубулопатию педиатр назначает комплексное исследование мочи и функций мочевыделительной системы. Обязательным является определение уровня калия, кальция, натрия, хлора в моче, а также тест на наличии глюкозы в моче. Обнаружить аминокислоты в анализе можно с помощью хроматографии. Конкременты исследуются визуально под микроскопом или выявляются при рентгенографическом обследовании и УЗИ. Кроме того, УЗИ-диагностика часто подтверждает аномалии развития почек, мочеточников и других отделов мочевыделительной системы. Для многих первичных тубулопатий установлен и тип наследования, и локализация дефекта в хромосоме, поэтому необходим анализ родословной. При возможности применяются молекулярно-генетические методы.
Содержание различных микроэлементов устанавливается не только в моче, но и в крови. Как правило, при повышенной экскреции почками в крови отмечается их дефицит. Также биохимический анализ крови позволяет оценить уровень глюкозы. Наряду с этим при тубулопатиях определяется количественное содержание гормонов. Особое внимание уделяется гормонам щитовидной железы и надпочечников, принимающим активное участие в метаболизме натрия, калия, кальция и т. д. Кроме того, высокая концентрация, например, альдостерона, может свидетельствовать об отсутствии чувствительности к нему рецепторов на мембранах тубулоцитов, что позволяет заподозрить почечный солевой диабет.
Лечений тубулопатий
Поскольку патология чаще всего обусловлена генетическими дефектами, этиотропная терапия не разработана. Исключение составляют вторичные тубулопатии, развившиеся в результате болезней накопления, нерациональной диеты и т. п. В таких случаях коррекция предрасполагающих факторов приводит к улучшению состояния. Например, при ожогах тяжелой степени проводится гемодиализ, при лизосомных болезнях накопления показана заместительная ферментная терапия, предупреждающая отложение различных соединений в почечных канальцах. Независимо от причины тубулопатий назначается ощелачивающая диета, включающая картофель, капусту, молочные продукты и фрукты.
Терапия первичных тубулопатий направлена на нормализацию уровня микроэлементов, которые усиленно выводятся с мочой. Острый дефицит корригируется внутривенным введением раствора хлорида натрия (чаще гипертонического), препаратов калия и цитратных смесей. В лечении рахита используются высокие дозы витамина D и бисфосфонаты, способствующие задержке кальция и фосфора в организме. Выраженные костные деформации являются показанием к хирургической коррекции. Поскольку при тубулопатиях часто отмечается задержка физического развития, возможно назначение курсов синтетического соматотропина.
Прогноз и профилактика тубулопатий
Прогноз зависит от причины развития заболевания. Почечный несахарный диабет у детей раннего возраста часто приводит к летальному исходу в результате быстрого обезвоживания. При болезни де Тони-Дебре-Фанкони в короткие сроки развивается почечная недостаточность, требующая заместительной терапии. При этом многие тубулопатии с нарушением мембранного транспорта аминокислот имеют более благоприятный прогноз, а почечная глюкозурия легкой степени вообще может протекать бессимптомно. В целом при первичных тубулопатиях прогноз хуже, чем при вторичных, поскольку отсутствует возможность этиотропной терапии. Профилактика заключается в медико-генетическом консультировании будущих родителей.
Нарушения, развивающиеся в результате дисфункции почечных канальцев (педиатрия)
Общая информация
Краткое описание
Нарушения, развивающиеся в результате дисфункции почечных канальцев (тубулопатии) – группа заболеваний, в основе которых лежат нарушения процессов канальцевого транспорта органических веществ или электролитов [1].

Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран

Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
Диагностика
II. МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ
рост, см х коэффициент
креатинин крови, мкмоль/л
— постпубертатный период 48-62
• УЗДГ сосудов почек.
• физикальный осмотр (определение ЧД, ЧСС).
• в семейном анамнезе – наличие родственников с ХБП, ХПН и/или с костными деформациями.
Возможны варианты сочетаний вышеуказанных симптомов.
Показания для консультации узких специалистов (с указанием цели консультации):
Дифференциальный диагноз
| Признаки | Антенатальный синдром Барттера | Антенатальный синдром Барттера с тугоухостью | Классическим синдром Барттера | Синдром Гиттельмана |
| Генный дефект | SLC12A1 KCN1 | BSND двугенная | CLCNKB | SLC1A3 |
| Возраст развития симптомов | антенатально | антенатально | варьирует | Детский и подростковый возраст |
| Есть | Есть | Редко | Нет | |
| Полиурия | Есть | Есть | Обычно есть | Нет |
| Остановка в развитии | Есть | Есть | Обычно есть | Нет |
| Отставание в росте | Есть | Есть | Обычно есть | Очень редко |
| Мышечные спазмы, тетания, слабость | Есть | Нет | Обычно есть | Есть |
| Нефрокальциноз | Есть | Нет | Редко | Нет |
| Нейросенсорная тугоухость | Есть | Есть | Нет | Нет |
| Эпизоды дегидратации | тяжелые | тяжелые | Редко | легкие |
| Магний в плазме | нормальный | Нормальный или низкий | Нормальный или низкий | легкие |
| Экскреция кальция с мочой | высокая | Транзиторно низкая или нормальная | Обычно нормальная низкая | низкий |
| Экскреция хлорида натрия с мочой | высокая | Очень высокая | Вариабельное повышение | Слегка повышена |
| Максимальная осмолярность мочи | Изо-/гипостенурия | Изо-/гипостенурия | Обычно нормальная | нормальная |
| Тип | Наследование | Клинические и биохимические признаки | Молекулярная генетика |
| I | АРТ | Мутации в трех генах, кодирующих субъединицы эпителиального натриевого канала (ENAC) | |
| АДТ | |||
| II | АД | Синдром Gordon: гиперкалиемия, гипертензия, гиперхлоремический ацидоз, нормальный АП, низкая АРП | WNK4, WNK1 |
| III | Приобретенный | Гиперкалиемия, ацидоз, повышение АП и АРП, снижение СКФ. | Является вторичной по отношению к обструктивным уропатиям, серповидно-клеточной и свинцовой нефропатии, амилоидозу, тяжелому пиелонефриту. |
Дифференциальная диагностика наследственных типов рахита, протекающих с дисфункциями почечных канальцев, представлена в таблице 4.
Нефрогенный несахарный диабет возникает у мальчиков на 1-м году жизни, характерны полиурия, полидипсия, с эпизодами гипернатриемической дегидратации, лихорадкой, раздражительностью и рвотой, задержкой развития. У девочек – дебют позже и протекает легче.
Тубулопатии у детей. Клинические рекомендации.
Тубулопатии у детей
Оглавление
Ключевые слова
Список сокращений
УЗИ – ультразвуковое исследование
РТА – ренальный тубулярный ацидоз
КЩС – кислотно-щелочное состояние
АДГ – антидиуретический гормон
УЗИ – ультразвуковое исследование
Термины и определения
Новые и узконаправленные профессиональные термины в настоящих клинических рекомендациях не используются.
1. Краткая информация
1.1 Определение
Тубулопатии – канальцевые болезни почек, характеризуемые различными нарушениями тубулярного транспорта электролитов, минералов, воды и органических субстанций, наследственного (первичные тубулопатии) или приобретенного характера (вторичные тубулопатии).
По локализации транспортного дефекта различают проксимальные, петлевые и дистальные тубулопатии.
1.2 Этиология и патогенез
1.2.1 Проксимальные тубулопатии
1.2.1.1 Гипофосфатемический рахит
Гипофосфатемический рахит (фосфат-диабет) – заболевание, связанное с дефектом реабсорбции фосфатов в проксимальных канальцах, проявляющееся у детей фосфатурией, гипофосфатемией и выраженными рахитическими изменениями, резистентными к обычным дозам витамина D [1, 5, 17, 18].
Описано несколько наследственных форм болезни, протекающих с изолированным нарушением проксимальной реабсорбции фосфатов в почках [1, 5, 17]:
FGF23 способствует развитию фосфатурии посредством угнетения реабсорбции фосфатов, вследствие редукции NPT2а, NPT2c и подавления экспрессии 1-?-гидроксилазы с последующей супрессией циркулирующего 1,25(OH)2D.
Паратгормон так же ингибирует реабсорбцию фосфатов в проксимальных канальцах, инактивируя натрий-фосфатные котранспортеры, но в отличие от FGF23 одновременно индуцирует транскрипцию 1-?-гидроксилазы, стимулируя синтез 1,25(OH)2D в проксимальных канальцах, что ведет к повышению NPT2b-зависимой кишечной абсорбции фосфатов и подавлению транскрипции генов паратгормона.
При Х-сцепленном доминантном гипофосфатемическом рахите мутации в гене фосфат-регулирующей гомологичной эндопептидазы в локусе Хр22.1 (PHEX – phosphate-regulating endopeptidase homolog, X-linked), приводят к нарушению ферментных систем, осуществляющих протеолиз FGF23. Избыток FGF23 обуславливает нарушение реабсорбции фосфатов в проксимальных канальцах почек, что формирует характерный биохимический фенотип, проявляющийся фосфатурией, гипофосфатемией, низким или нормальным, но неадекватно сниженным относительно гипофосфатемии уровнем 1,25(OH)2D3. Несмотря на то, что к настоящему времени описано более 170 мутаций PHEX-гена (миссенс, нонсенс, делеции, сплайс-сайт мутации), отчетливые генотип-фенотипические корреляции не описываются.
Причиной аутосомно-доминантного гипофосфатемического рахита является непосредственное возникновение мутаций в гене FGF23 на хромосоме 12p13.3, формирующих устойчивость фактора к протеолитическому расщеплению.
Возникновению аутосомно-рецессивного гипофосфатемического рахита способствуют мутации в гене дентин матриксного протеина 1 (dentin matrix protein 1 – DMP1) на хромосоме 4q21 или гене эктонуклеоид пирофосфатазы/фосфодиэстеразы 1 (endonucleotide pyrophosphatase/phosphodiesterase 1 – ENPP1) на хромосоме 6q22-q23, так же способствующие повышению концентраций FGF23.
Аутосомно-рецессивный наследственный гипофосфатемический рахит с гиперкальциурией развивается вследствие мутаций в гене SLC4A3 на хромосоме 9q34, непосредственно кодирующем натрий-фосфатный котранспортер (NPT2c) люминальной мембраны проксимальных канальцев.
1.2.1.2 Проксимальный ренальный тубулярный ацидоз
Проксимальный РТА (II тип) (OMIM 179830) – заболевание, характеризующееся нарушением реабсорбции бикарбонатов (НСО3-) в проксимальных канальцах [1, 2, 11, 12].
Первичный проксимальный РТА (изолированный) [1, 2, 11, 12]:
Вторичный проксимальный РТА обусловлен рядом заболеваний: цистиноз, галактоземия, гликогеноз (тип I), тирозинемия, болезнь Вильсона, гиперпаратиреоидизм, медуллярная кистозная болезнь, витамин-Д-дефицитный и зависимый рахит, идиопатическая гиперкальциурия, первичная гипероксалурия, синдром Лоу, синдром Шегрена, множественная миелома. Также может быть вызван токсическим поражением проксимальных канальцев солями тяжелых металлов, некоторыми лекарственными препаратами.
1.2.1.3 Синдром Фанкони
Синдром Фанкони (де Тони-Дебре) – заболевание, обусловленное генерализованной дисфункцией проксимальных канальцев, приводящей к нарушению реабсорбции аминокислот, глюкозы, калия, натрия, воды, фосфатов, бикарбонатов, мочевой кислоты [1, 2, 11].
Различают две формы заболевания [1, 2, 7, 8, 11, 25]:
Наиболее частой причиной синдрома Фанкони у детей является цистиноз (OMIM 219800) [1,7,8], редкое аутосомно-рецессивное заболевание, которое характеризуется накоплением кристаллов цистина внутри лизосом и сопровождается прогрессирующим поражением интерстициальной ткани почек с исходом в хроническую почечную недостаточность; частота встречаемости
1:200000 новорожденных (Европа, США).
Дефект лизосомального переносчика цистина – цистинозина вызывается различными мутациями в гене CTNS (хромосома 17p13). Наиболее часто выявляемая большая делеция гена СTNS полностью нарушает его функцию.
Нарушение транспорта цистина через лизосомальную мембрану ведет к накоплению цистина в лизосоме, снижению цистина и цистеина в цитозоле, что приводит к повышению продукции реактивных радикалов кислорода, вызывает истощение АТФ и стимулирует апоптоз.
1.2.1.4 Ренальная глюкозурия
Ренальная глюкозурия – заболевание, обусловленное нарушением транспорта глюкозы в проксимальных канальцах почек, при нормальном уровне глюкозы в крови [20, 23].
В физиологических условиях глюкоза полностью реабсорбируется в проксимальных канальцах. Реабсорбция основной массы глюкозы происходит в S1 и S2 сегментах при участии почечно-специфичного натрий-глюкозного транспортёра-2 люминальной мембраны. Оставшаяся часть глюкозы удаляется из фильтрата в S3 сегменте посредством натрий-глюкозного транспортёра-1. Этот транспортёр так же присутствует и в тонкой кишке. Как и другие мембранные транспортные системы, транспортёры глюкозы имеют предел насыщаемости. При снижении почечного порога для глюкозы, несмотря на нормальный уровень сахара в крови, появляется ренальная глюкозурия.
1.2.2 Петлевые тубулопатии
1.2.2.1 Синдром Барттера
Синдром Барттера – аутосомно-рецессивное заболевание, обусловленное дефектом реабсорбции натрия и хлоридов в толстом восходящем колене петли Генле, для которого характерно развитие гипокалиемии, гипохлоремии, метаболического алкалоза и гиперренинемического гиперальдостеронизма [1, 9, 14, 21].
При неонатальном варианте синдрома Барттера развивается гиперкальциурия и нефрокальциноз.
Классический вариант (тип III) сопровождается нарушением транспорта хлоридов через базолатеральную мембрану обратно в циркуляцию, что ведёт к гиповолемии и последующей активации ренин-ангиотензиновой системы с развитием гипокалиемического метаболического алкалоза.
1.2.3 Дистальные тубулопатии
1.2.3.1 Синдром Гительмана
Синдром Гительмана (OMIM 263800), семейная гипокалиемическая гипомагнеземия, сольтеряющая тубулопатия, характеризующаяся гипомагнеземией, гипокальциурией и вторичным альдостеронизмом, который приводит к развитию гипокалиемии и метаболическому алкалозу [1, 9, 13].
1.2.3.2 Дистальный ренальный тубулярный ацидоз (I тип)
Дистальный РТА (I тип) (OMIM 179800, OMIM 602722) – заболевание, характеризующееся тяжелым гиперхлоремическим метаболическим ацидозом, вследствие нарушения экскреции водородных ионов в дистальном отделе нефрона [1, 2, 4].
Первичный дистальный РТА:
Наряду с семейными формами заболевания, встречаются и спорадические случаи.
Вторичные (приобретённые) формы дистального РТА описаны при многих патологических состояниях, обусловленных расстройствами метаболизма кальция с нефрокальцинозом и гиперкальциурией, первичным гиперпаратиреоидизмом, лекарственным и токсическим повреждением, другими почечными заболеваниями, в том числе медуллярной кистозной болезнью и губчатой почкой, аутоиммунными заболеваниями (гипергаммаглобулинемия, синдром Шегрена, аутоиммунный гепатит, тиреоидит, фиброзирующий альвеолит, системная красная волчанка, узелковый периартериит).
Нарушение экскреции аммония при всех вариантах вторично. Реабсорбция бикарбоната количественно нормальна, но, в соответствии с повышением рН мочи, определенная степень бикарбонатурии обязательно присутствует (менее 5% профильтрованного количества).
При тяжелом хроническом метаболическом ацидозе кость обеспечивает до 40% буферной емкости крови; нейтрализация ионов водорода костным карбонатом вызывает высвобождение кальция из кости во внеклеточную жидкость, что ведёт к нарушению её нормальной структуры и разнообразным костным деформациям.
Экскреция цитрата в проксимальном канальце снижена, что является основой формирования нефрокальциноза.
1.2.3.3 Псевдогипоальдостеронизм
У младенцев описан транзиторный синдром гиперкалиемии и метаболического ацидоза без потери соли, его рассматривают как вариант ренального псевдогипоальдостеронизма тип I.
Способность подкислять мочу после нагрузки кислотами при заболевании сохранена, но экскреция кислот почками снижена в связи с очень низкой экскрецией аммония. Хотя снижение продукции аммония обусловлено самой гиперкалиемией, в механизме развития этой формы ключевую роль играет дефицит альдостерона или резистентность к нему почечных канальцев.
Повышена реабсорбция хлорида натрия в толстом восходящем колене петли Генле, что приводит к нарушению секреции калия и водорода в кортикальных собирательных трубочках.
1.2.3.4 Нефрогенный несахарный диабет
Х-сцепленная рецессивная форма (OMIM 304800) – мутации гена AVPR2 (локус Хq28), кодирующего рецептор к аргинин-вазопрессину (V2R) в клетках собирательных трубочек.
Активируясь при связывании с вазопрессином, рецептор V2 вызывает повышение цАМФ. Это приводит к передвижению в сторону апикальной мембраны внутриклеточных везикул содержащих водные каналы аквапорина-2 (AQ-2), что повышает проницаемость канальцев для воды. Генетический дефект, включающий различное количество мутаций в гене V2-рецептора, приводит к снижению связывания вазопрессина с рецептором, уменьшению синтеза или увеличению деградации самого рецептора. В результате блокируется антидиуретическое действие антидиуретического гормона (АДГ). Различные мутации ассоциированы с вариабельной резистентностью к АДГ. Х-сцепленный вариант наследования подразумевает наличие выраженной полиурии у мальчиков; у асимптоматичных большую часть времени лиц женского пола полиурия может возникнуть в течение беременности, когда секреция плацентарной вазопрессиназы приводит к повышению клиренса эндогенного АДГ.
Аутосомно-рецессивная форма (OMIM 125800) вызвана мутациями в гене аквапорина-2 (AQP2, локус 12q13). Болеют как мальчики, так и девочки.
Пострецепторный дефект заключён в нарушении движения и последующего слияния с люминальной мембраной АДГ-чувствительных водных каналов аквапорина-2, локализованных в цитозоле главных клеток собирательных трубок, что препятствует пассивной диффузии воды.
Существование аутосомно-доминантной формы наследования болезни оспаривается, но единичные случаи описаны.
Помимо генетически-детерминированных, встречаются и спорадические случаи болезни.
1.2.3.5 Синдром Лиддла
Синдром Лиддла (псевдоальдостеронизм) (OMIM 177200) – наследственное заболевание, характеризующееся ранним дебютом тяжелой артериальной гипертензии, в сочетании с низкими уровнями активности ренина и альдостерона плазмы, гипокалиемией и метаболическим алкалозом [10].
Замедление деградации ENaC проявляется избыточной реабсорбцией натрия и потерей калия. Избыточная реабсорбция натрия ведет к артериальной гипертензии вследствие увеличения объема циркулирующей крови, что подавляет секрецию ренина и альдостерона.
1.3 Эпидемиология
Изолированный проксимальный РТА, первичный генетически-детерминированный синдром Фанкони (OMIM 134600, OMIM 615605, OMIM 613388) встречаются крайне редко. Данные о распространенности заболеваний отсутствуют.
Показатели распространенности семейной ренальной глюкозурии варьируют от 0,6% до 6,3%, в зависимости от диагностических критериев, используемых для постановки диагноза.
Изолированный дистальный РТА встречается крайне редко. Данные о распространенности заболевания отсутствуют.
Псевдогипоальдостеронизм встречается крайне редко, частота встречаемости 1:47000 живых новорожденных.
Эпидемиология нефрогенного несахарного диабета зависит от типа наследования. Х-сцепленный нефрогенный несахарный диабет крайне редкое заболевание, тем не менее, по частоте встречаемости превышает аутосомно-рецессивный тип в соотношении 9:1. Частота мутаций у лиц мужского пола составляет 4 случая на 1000.000 населения.
Синдром Лиддла встречается крайне редко. Данные о распространенности заболевания отсутствуют.
1.4 Кодирование по МКБ-10
Нарушения, развивающиеся в результате дисфункции почечных канальцев (N25):
N25.0 – Почечная остеодистрофия;
N25.1 – Нефрогенный несахарный диабет;
N25.8 – Другие нарушения, обусловленные дисфункцией почечных канальцев;
N25.9 – Нарушение функции почечных канальцев уточненное.
1.5 Примеры диагнозов
1.6 Классификация
По локализации транспортного дефекта различают проксимальные, петлевые и дистальные тубулопатии.
К проксимальным тубулопатиям относят гипофосфатемический рахит проксимальный РТА, синдром Фанкони ренальную глюкозурию.
К петлевым тубулопатиям относят синдром Барттера (различают неонатальный и классический синдром Барттера).
К дистальным тубулопатиям относят синдром Гительмана, дистальный РТА, псевдогипоальдостеронизм, нефрогенный несахарный диабет, синдром Лиддла.
2. Диагностика
2.1 Жалобы и анамнез
2.1.1 Проксимальные тубулопатии
2.1.1.1 Гипофосфатемический рахит
Важен анализ родословной: поиск случаев подтверждённого заболевания у членов семьи и родственников.
Заболевание манифестирует в возрасте 9-13 месяцев. Клиническая картина проявляется отставанием в росте, разнообразными симптомами рахита (наиболее ранний симптом – прогрессирующее, несмотря на проведение профилактики рахита обычными дозами витамина D, искривление нижних конечностей). Зубы появляются с опозданием, типичны дефекты эмали и множественный кариес.
2.1.1.2 Проксимальный ренальный тубулярный ацидоз (II тип)
Наиболее часто заболевание манифестирует в возрасте 1–18 мес. Единственное клиническое проявление аутосомно-доминантного типа – отставание в росте [1, 2, 11, 12]. При аутосомно-рецессивном типе наряду с отставанием в росте присутствуют глазные аномалии (глаукома, катаракта), а также отставание в умственном развитии [1, 2, 11]. Для транзиторного младенческого типа характерны: задержка роста, снижение аппетита, тошнота, рвота, приступы дегидратации и гипотонии.
2.1.1.3 Синдром Фанкони (де Тони-Дебре)
Симптомы синдрома Фанкони: полиурия, дегидратация, мышечная слабость, отсутствие аппетита, плохая прибавка в весе, задержка роста, рахитоподобные изменения скелета, отставание в умственном развитии.
Наиболее частой причиной синдрома Фанкони у детей является цистиноз. Инфантильная форма нефропатического цистиноза манифестирует с синдрома Фанкони в возрасте 6-12 месяцев с быстрой прогрессией до терминальной стадии хронической почечной недостаточности (к 8-12 годам) [1, 7, 8].
Ювенильную форму нефропатического цистиноза отличают более поздний дебют в течение пубертатного периода, меньшая выраженность клиники синдрома Фанкони, медленная прогрессия до хронической почечной недостаточности. Взрослая форма болезни протекает с изолированным поражением глаз [1, 7, 8].
2.1.1.4 Ренальная глюкозурия
Изолированная ренальная глюкозурия (тип А) протекает бессимптомно.
Основными клиническими симптомами глюкозо-галактозной мальабсорбции (тип В) являются: водянистая диарея, отсутствие прибавки в весе, признаки обезвоживания.
2.1.2 Петлевые тубулопатии
2.1.2.1 Синдром Барттера
2.1.3 Дистальные тубулопатии
2.1.3.1 Синдром Гительмана
В течение длительного времени заболевание может протекать бессимптомно, изредка наблюдаются эпизоды лихорадки, рвоты, болей в животе, мышечной слабости, тетании.
2.1.3.2 Дистальный ренальный тубулярный ацидоз (I тип)
2.1.3.3 Псевдогипоальдостеронизм
Псевдогипоальдостеронизм тип I включает две клинические формы: с ренальными и с полиорганными нарушениями.
Наиболее часто наблюдают ренальную аутосомно-доминантную форму. Клиника вариабельна, может протекать с угрозой жизни в связи с тяжелой потерей соли и выраженной гиперкалиемией.
В возрасте 1-2 лет может наступить улучшение, предположительно, за счет «дозревания» проксимального тубулярного транспорта, развития «солевого» аппетита и улучшения ренального тубулярного ответа на минералокортикоиды.
При второй форме с множественной органной резистентностью к минералокортикоидам эпизоды потери соли отмечаются сразу после рождения, тогда же возможен и летальный исход.
У младенцев также описан транзиторный синдром гиперкалиемии и метаболического ацидоза без потери соли. Эта форма названа гиперкалиемией раннего возраста, её рассматривают как вариант ренального псевдогипоальдостеронизма тип I.
2.1.3.4 Нефрогенный несахарный диабет
Первые признаки болезни наблюдают в возрасте 3-6 мес:
В раннем возрасте полиурия и полидипсия маскируются физиологической полиурией и полидипсией грудного возраста, так же может наблюдаться отсутствие жажды, в связи с незрелостью её центра или нечуствительностью осморецепторов.
В возрасте старше года жажда и полиурия ярко выражены, дети выпивают и выделяют до 6-10 л/(м 2 /сут). В дальнейшем может присоединиться задержка психомоторного развития, степень её зависит от времени постановки диагноза. При рано начатом лечении отставание можно ликвидировать.
2.1.3.5 Синдром Лиддла
Характерны: тяжелая артериальная гипертензия, быстрая утомляемость, полиурия мышечная гипотония.
2.2 Физикальное обследование
2.2.1 Проксимальные тубулопатии
2.2.1.1 Гипофосфатемический рахит (фосфат-диабет)
Общий осмотр подразумевает оценку физического развития ребенка, состояния костной системы, подсчет частоты дыхания, сердечных сокращений, аускультацию легких, сердца, пальпацию живота.
2.2.1.2 Проксимальный ренальный тубулярный ацидоз (II тип)
Общий осмотр подразумевает оценку физического развития ребенка, подсчет частоты дыхания, сердечных сокращений, аускультацию легких, сердца, пальпацию живота.
Характерен низкий рост.
2.2.1.3 Синдром Фанкони (де Тони-Дебре)
Общий осмотр подразумевает оценку физического развития ребенка, умственного развития ребенка, состояния костной системы, мышечной системы, подсчет частоты дыхания, сердечных сокращений, аускультацию легких, сердца, пальпацию живота, учет объема выпитой жидкости, диуреза.
Характерны [1, 7, 8, 11]:
Ранними и патогномоничными экстраренальными проявлениями нефропатического цистиноза считаются отложения кристаллов цистина в роговице (кератопатия), выявляющиеся со второго года жизни, в дальнейшем могут поражаться эндокринные органы (гипотиреоз, сахарный диабет, гипогонадизм (у мальчиков), нервная система (нейромиопатия, эпилепсия, мозжечковые и пирамидные расстройства, отставание в умственном развитии), печень и поджелудочная железа [7, 8].
2.2.1.4 Ренальная глюкозурия
Общий осмотр подразумевает оценку физического развития ребенка, состояния мышечной системы, подсчет частоты дыхания, сердечных сокращений, аускультацию легких, сердца, пальпацию живота, учет объема выпитой жидкости, диуреза, исследование частоты и характера стула.
Для глюкозо—галактозной мальабсорбции (тип В) характерны:
2.2.2 Петлевые тубулопатии
2.2.2.1 Синдром Барттера
Общий осмотр подразумевает оценку физического развития ребенка, мышечной системы, подсчет частоты дыхания, сердечных сокращений, аускультацию легких, сердца, пальпацию живота, учет объема выпитой жидкости, диуреза.
2.2.3 Дистальные тубулопатии
2.2.3.1 Синдром Гительмана
В течение длительного времени заболевание может протекать бессимптомно. Общий осмотр подразумевает оценку физического развития ребенка, состояния мышечной системы, подсчет частоты дыхания, сердечных сокращений, аускультацию легких, сердца, пальпацию живота, учет объема выпитой жидкости, диуреза.
2.2.3.2 Дистальный ренальный тубулярный ацидоз (I тип)
Общий осмотр подразумевает оценку физического развития ребенка, состояния костной системы, мышечной системы, подсчет частоты дыхания, сердечных сокращений, аускультацию легких, сердца, пальпацию живота, учет объема выпитой жидкости, диуреза.
2.2.3.3 Псевдогипоальдостеронизм
Общий осмотр подразумевает оценку физического развития ребенка, измерение артериального давления, подсчет частоты дыхания, сердечных сокращений, аускультацию легких, сердца, пальпацию живота.
Псевдогипоальдостеронизм тип I: для обеих форм (ренальной и полиорганной) характерны задержка физического развития, симптомы дегидратации, артериальная гипотензия. Для полиорганной формые (сразу после рождения), помимо симптомов дегидратации, также характерны затруднение дыхания, кашель, одышка.
Псевдогипоальдостеронизм тип II (синдром Gordon) развивается в подростковом возрасте, всегда присутствует тяжелая артериальная гипертензия.
2.2.3.4 Нефрогенный несахарный диабет
Общий осмотр подразумевает оценку физического развития ребенка, измерение артериального давления, подсчет частоты дыхания, сердечных сокращений, аускультацию легких, сердца, пальпацию живота. Характерны: низкое/очень низкое физическое развитие, симптомы дегидратации, полиурия, полидипсия.
2.2.3.5 Синдром Лиддла
Общий осмотр подразумевает оценку физического развития ребенка, измерение артериального давления, подсчет частоты дыхания, сердечных сокращений, аускультацию легких, сердца, пальпацию живота.
2.3 Лабораторная диагностика
2.3.1 Проксимальные тубулопатии
2.3.1.1 Гипофосфатемический рахит
Ведущими лабораторными симптомами FGF23-зависимых форм гипофосфатемического рахита (Х-сцепленный доминантный ГФР, аутосомно-доминантный ГФР, аутосомно-рецессивный ГФР) являются: гипофосфатемия (менее 0,8 ммоль/л), фосфатурия [1, 5, 17]. Кальций сыворотки и 25(OH)D3 в норме, уровень 1,25(OH)2D3 низкий или нормальный, уровень паратгормона нормальный или незначительно повышен [1, 5, 17]. Отсутствует метаболический ацидоз. Повышена активность щелочной фосфатазы. Почечные функции остаются сохранными.
(Сила рекомендаций 3; уровень доказательств C)
Комментарий: Диагностические критерии FGF23-зависимых форм гипофосфатемического рахита представлены в таблице 1.
Таблица 1. Диагностические критерии FGF23-зависимых форм гипофосфатемического рахита
Показатель
X-ГФР
Фосфор сыворотки
Ниже нормы (гипофосфатемия)
Экскреция фосфатов с мочой
Выше нормы (фракционная экскреция > 15%)
Экскреция кальция с мочой
Экскреция белка с мочой
Экскреция аминокислот с мочой
Экскреция глюкозы с мочой
Отсутствие метаболического ацидоза
2.3.1.2 Проксимальный ренальный тубулярный ацидоз (II тип)
(Сила рекомендаций 3; уровень доказательств С)
Комментарии:
Диагностические критерии проксимального ренального тубулярного ацидоза (II тип)
2.3.1.3 Синдром Фанкони (де Тони-Дебре)
(Сила рекомендаций 3; уровень доказательств С)
Комментарии:
Критерии диагностикисиндрома Фанкони [1, 7, 8, 11]:
2.3.1.4 Ренальная глюкозурия
(Сила рекомендаций 3; уровень доказательств С)
Комментарии:
Диагностические критерии ренальной глюкозурии:
2.3.2 Петлевые тубулопатии
2.3.2.1 Синдром Барттера
(Сила рекомендаций 3; уровень доказательств С)
Комментарии:
Диагностические критерии синдрома Барттера:
При классическом варианте синдрома нефрокальциноз отсутствует [1, 9, 21].
2.3.3 Дистальные тубулопатии
2.3.3.1 Синдром Гительмана
(Сила рекомендаций 3; уровень доказательств C)
Комментарии:
Диагностические критерии синдрома Гительмана:
2.3.3.2 Дистальный ренальный тубулярный ацидоз (I тип)
(Сила рекомендаций 3; уровень доказательств C)
Комментарии:
Диагностические критерии дистального РТА (1 тип):
2.3.3.3 Псевдогипоальдостеронизм
(Сила рекомендаций 4; уровень доказательств C)
Комментарии:
Диагностические критерии псевдогипоальдостеронизма:
Псевдогипоальдостеронизм тип I
Псевдогипоальдостеронизм тип II (синдром Gordon)
2.3.3.4 Нефрогенный несахарный диабет
(Сила рекомендаций 3; уровень доказательств C)
Комментарии:
Проба с экзогенным антидиуретическим гормоном
ДДАВП-тест (1-дезамино-8-D-аргинин вазопрессин-тест) – проба с введением АДГ. Суть пробы заключена в определении реакции почечного концентрационного механизма на введение экзогенного вазопрессина. Детям до года данную пробу проводят в исключительных случаях.
Осмоляльность мочи после применения препарата должна повышаться до 800-900 мОсм/кг (плотность до 1020-1025) в последовательно собранных анализах мочи. Отсутствие повышения осмоляльности и относительной плотности мочи подтверждает резистентость собирательных трубочек к действию АДГ, что характерно для нефрогенного несахарного диабета.
Диагностические критерии нефрогенного несахарного диабета
Исследование крови:
2.3.3.5 Синдром Лиддла
(Сила рекомендаций 4; уровень доказательств C)
Комментарии:
Диагностические критерии синдрома Лиддла
2.4 Инструментальная диагностика
2.4.1 Проксимальные тубулопатии
2.4.1.1 Гипофосфатемический рахит
(Сила рекомендаций 3; уровень доказательств C)
(Сила рекомендации 3; уровень доказательств C)
2.4.1.2 Проксимальный ренальный тубулярный ацидоз (II тип)
(Сила рекомендаций 3; уровень доказательств С)
Комментарий: При первичном проксимальном ренальном тубулярном ацидозе УЗИ почек всегда в норме.
2.4.1.3 Синдром Фанкони (де Тони-Дебре)
Визуализационные исследования не играют важной роли в постановке собственно диагноза синдрома Фанкони (де Тони-Дебре).
(Сила рекомендаций 3; уровень доказательств С).
2.4.1.4 Ренальная глюкозурия
Комментарий: Визуализационные исследования не требуются при изолированной ренальной глюкозурии (тип А) [20, 23].
(Сила рекомендации 3; уровень доказательств С)
2.4.2 Петлевые тубулопатии
2.4.2.1 Синдром Барттера
(Сила рекомендации 3; уровень доказательств С)
2.4.3 Дистальные тубулопатии
2.4.3.1 Синдром Гительмана
(Сила рекомендации 4; уровень доказательств С)
2.4.3.2 Дистальный ренальный тубулярный ацидоз (I тип)
(Сила рекомендации 3; уровень доказательств C)
(Сила рекомендации 3; уровень доказательств C)
2.4.3.3 Псевдогипоальдостеронизм
(Сила рекомендации 3; уровень доказательств C)
2.4.3.4 Нефрогенный несахарный диабет
(Сила рекомендации 3; уровень доказательств C)
2.4.3.5 Синдром Лиддла
Комментарий: Визуализационные исследования при синдроме Лиддла неинформативны [10].
(Сила рекомендации 4; уровень доказательств C)
2.5 Иная диагностика
2.5.1 Проксимальные тубулопатии
2.5.1.1 Гипофосфатемический рахит
(Сила рекомендации 3; уровень доказательств С)
2.5.1.2 Проксимальный ренальный тубулярный ацидоз (II тип)
(Сила рекомендации 4; уровень доказательств С)
2.5.1.3 Синдром Фанкони (де Тони-Дебре)
(Сила рекомендации 3; уровень доказательств С)
Комментарий: при офтальмологическом обследовании можно обнаружить отложения кристаллов цистина в роговице, колец Кайзера-Флейшера. Также оправдана консультация офтальмолога при подозрении на галактоземию, синдром Лоу (врожденная катаракта).
(Сила рекомендации 3; уровень доказательств С)
Комментарии:
Критерии диагностики нефропатического цистиноза, как причины синдрома Фанкони [1, 7, 8]:
2.6.3.5 Синдром Лиддла
3. Лечение
3.1 Консервативное лечение
3.1.1 Проксимальные тубулопатии
3.1.1.1 Гипофосфатемический рахит
Стандартная терапия FGF23-медиируемых гипофосфатемических рахитов (Х-ГФР, аутосомно-доминантный ГФР, аутосомно-рецессивный ГФР) основана на одновременном назначении неорганических фосфатов и активных аналогов витамина D (чаще кальцитриол ж,вк ; так же используется альфакальцидол ж,вк ) и направлена на клиническое излечение рахита, улучшение гистологии костной ткани [1, 5, 6, 18]. Раннее начало лечения позволяет избежать деформации костей. Дозы и длительность лечения определяются выраженностью рахитических изменений, уровнем фосфатов в крови, возрастом пациентов. Более высокие дозы препаратов необходимы в начале терапии и в периоды интенсивного роста ребенка.
(Сила рекомендаций 3; уровень доказательств C)
Комментарий: Возможно повышение дозы фосфатов в периоды интенсивного роста (до 55-70 мг/кг в день по элементарному фосфору). Цель – достижение уровня фосфатов сыворотки 1,0-1,2 ммоль/л.
В настоящее время применяется раствор неорганических фосфатов (однозамещенный фосфат натрия 2-водный – 5 г и двухзамещенный фосфат натрия 12-водный – 10 г на 250 мл воды), конечный раствор содержит 7,44 мг элементарного фосфора в 1 мл.
В большинстве случаев не удается полностью нормализовать уровень фосфатов в сыворотке.
Замещение фосфата предпочтительнее проводить в форме Phosphat Sandoz, так как в одной таблетке содержится 790 мг цитрата, предотвращающего развитие нефрокальциноза. Также возможен прием препарата Reducto-spezial (натрийдигидрогенфосфат и динатрий гидрогенфосфат). Однако, оба препарата на территории Российской Федерации не зарегистрированы.
Оптимальные дозы препаратов 1,25(OH)2D3 не определены. С целью предотвращения формирования нефрокальциноза необходим динамический контроль экскреции кальция с мочой, содержания кальция, фосфора, активности щелочной фосфатазы в сыворотке крови, УЗИ почек. В качестве альтернативы кальцитриолу может быть использован альфакальцидол.
Роль лечения рекомбинантным гормоном роста оценивается противоречиво. Существует единственное рандомизированное исследование Zivicniak M. et al, которое показало значительное улучшение динамики роста у 8 из 16 детей, получавших рекомбинантный гормон роста [28].
3.1.1.2 Проксимальный РТА
(Сила рекомендаций 3; уровень доказательств C)
3.1.1.3 Синдром Фанкони (де Тони-Дебре)
При синдроме Фанкони основные лечебные мероприятия направлены на коррекцию канальцевых потерь жидкости и электролитов.
(Сила рекомендации 3; уровень доказательств C)
(Сила рекомендации 3; уровень доказательств C)
При недостаточной эффективности вышеперечисленных лечебных мероприятий по предотвращению потерь воды, калия, натрия рекомендовано применение индометацина 0,5-1,5 мг/кг/сут в два приема (обычно применяется до 2-летнего возраста) [1, 7, 8].
(Сила рекомендации 3; уровень доказательств C)
Комментарий: Применение препарата у детей off label – вне зарегистрированных в инструкции лекарственного средства показаний – осуществляется при наличии подписанного информированного согласия родителей / законных представителей (индометацин в виде таблеток официально разрешен к применению с 14 летнего возраста).
(Сила рекомендации 3; уровень доказательств С)
(Сила рекомендации 3; уровень доказательств С).
Специфическая терапия нефропатического цистиноза препаратом, содержащим цистеамина битартрат (не зарегистрирован в Российской Федерации) [1, 7, 8, 15, 16]. Лечение (для непролонгированной формы цистеамина битартрата) начинается с низкой дозы 0,2 г/м 2 /сут, при хорошей переносимости следует постепенно увеличивать дозу в течение 4-6 недель до целевой 1,30 г/м 2 (для пациентов до 12 лет) и 2 г/сут (для пациентов старше 12 лет, препарат необходимо принимать каждые 6 часов [1, 7, 8, 16]. Максимальная доза не должна превышать 1,95 г/м 2 /сут. (Сила рекомендаций 1b; уровень доказательств – B)
(Сила рекомендаций 3; уровень доказательств C)
(Сила рекомендаций 3; уровень доказательств C)
Комментарии: Применение индометацина у детей off label – вне зарегистрированных в инструкции лекарственного средства показаний – осуществляется при наличии подписанного информированного согласия родителей / законных представителей (индометацин в виде таблеток официально разрешен к применению с 14 летнего возраста).
3.1.3 Дистальные тубулопатии
3.1.3.1 Синдром Гительмана
(Сила рекомендаций 4; уровень доказательств C)
Комментарии: Высокие дозы магния способствуют возникновению диареи, поэтому нормализация уровней сывороточного магния является трудно достижимой задачей.
3.1.3.2 Дистальный РТА
Цель терапии – коррекция ацидоза, профилактика дальнейшего отложения кальция в почках.
(Сила рекомендаций 3; уровень доказательств C)
(Сила рекомендации 3; уровень доказательств C)
Комментарии: Возможна коррекция остеопатии активными метаболитами витамина D3 (индивидуальный подход, применяется с осторожностью из-за возможности усиления гиперкальциурии).
3.1.3.3 Псевдогипоальдостеронизм
(Сила рекомендаций 4; уровень доказательств C)
Комментарий: доза подбирается индивидуально, исходя из суточных потерь натрия с мочой, уровней натрия, калия в крови, показателей альдостерона и активности ренина в плазме. У детей первых 2 лет жизни дозы хлорида натрия, как правило, варьируют от 4 г/сут до 12 г/сут. В большинстве случаев с возрастом потребность в соли значительно снижается [1, 3].
(Сила рекомендации 4; уровень доказательств C).
(Сила рекомендаций 4; уровень доказательств C)
3.1.3.4 Нефрогенный несахарный диабет
(Сила рекомендации 3; уровень доказательств C)
(Сила рекомендаций 3; уровень доказательств C)
Комментарий: Пероральный прием жидкости не должен ограничиваться по количеству и времени.
(Сила рекомендации 3; уровень доказательств C)
Комментарии: Действие связано с индуцированным гиповолемией увеличением реабсорбции натрия и воды в проксимальном канальце, что снижает водную нагрузку на АДГ-чувствительные части собирательных трубок.
Возможно сочетанное применение двух препаратов, так как амилорид обладает и калийсберегающим действием [1, 26].
(Сила рекомендации 3; уровень доказательств C)
Комментарии: Блокируя ренальный синтез простагландинов, являющихся антагонистами антидиуретического гормона, препарат повышает концентрационную способность почек [1, 26, 27].
Применение препарата у детей off label – вне зарегистрированных в инструкции лекарственного средства показаний – осуществляется при наличии подписанного информированного согласия родителей / законных представителей (индометацин в виде таблеток официально разрешен к применению с 14 летнего возраста).
3.1.3.5 Синдром Лиддла
(Сила рекомендаций 4; уровень доказательств D)
Комментарий: применение спиронолактона нецелесообразно, в связи с резистентностью к нему [10].
3.2 Хирургическое лечение
(Сила рекомендации 3; уровень доказательств C)
Комментарий: хирургическая коррекция должна проводится после закрытия эпифизарных зон роста.
(Сила рекомендации 3; уровень доказательств C)
Комментарий: синдром Фанкони никогда не рецидивирует в трансплантате, а выявление кристаллов цистина в почечной ткани объясняется их заносом лейкоцитами и макрофагами хозяина. Родственники пациента, являющиеся гетерозиготными носителями цистиноза, могут быть донорами почки, т. к. это заболевание у них не развивается. Лечение цистеамином после трансплантации почки должно продолжаться для воздействия на экстраренальные проявления цистиноза.
4. Реабилитация
При тубулопатиях реабилитационные мероприятия не применяются.
Возможна реабилитация после ортопедической коррекции деформаций нижних конечностей у пациентов с гипофосфатемическим рахитом.
5. Профилактика и диспансерное наблюдение
5.1 Профилактика
Рекомендуется генетическое консультирование родителей детей с вторичным синдромом Фанкони, обусловленным генетическими болезнями (цистиноз, галактоземия, наследственная непереносимость фруктозы, тирозинемия (тип I), гликогеноз (тип I), болезнь Вильсона-Коновалова и др.), гипофосфатемическим рахитом, синдромом Барттера, синдромом Гительмана, нефрогенным несахарным диабетом, с целью объяснения закономерностей наследования и прогнозирования рисков повторения болезни при последующих беременностях.
Анализ родословной состоит в поиске случаев подтвержденного заболевания и его основных симптомов у членов семьи и родственников.
Вероятность рождения ребенка с повторным случаем синдрома Гительмана у родителей, уже имеющих больного ребенка, составляет 25%. Более старшим детям семьи, не имеющих признаков синдрома Гительмана, также необходимо проведение ДНК-диагностики, в связи с возможностью появления клинических симптомов в более поздней жизни. Учитывая хороший прогноз, антенатальный диагноз синдрома Гительмана не рекомендуется.
5.2. Ведение пациентов
5.2.1 Проксимальные тубулопатии
5.2.1.1 Гипофосфатемический рахит
5.2.1.2 Проксимальный ренальный тубулярный ацидоз (II тип)
5.2.1.3 Синдром Фанкони (де Тони-Дебре)
5.2.1.4 Ренальная глюкозурия
Наблюдение пациентов с изолированной ренальной глюкозурией не требуется.
5.2.2 Петлевые тубулопатии
5.2.2.1 Синдром Барттера
5.2.3 Дистальные тубулопатии
5.2.3.1 Синдром Гительмана
Амбулаторное наблюдение большинства бессимптомных пациентов осуществляется нефрологом 1-2 раза в год. В каждый визит пациента особое внимание уделяется жалобам, обусловленным гипокалиемией (усталость, мышечная слабость, запоры, сердечная аритмия) и гипомагнеземией (тетания, судороги, парестезии, боль в суставах и мышцах), определяются сывороточные уровни калия, магния, бикарбоната.
5.2.3.2 Дистальный ренальный тубулярный ацидоз (I тип)
5.2.3.3 Псевдогипоальдостеронизм
Контроль артериального давления, КЩС, уровня креатинина, электролитов крови (калий, натрий, хлориды).
5.2.3.4 Нефрогенный несахарный диабет
5.2.3.5 Синдром Лиддла
Контроль КЩС, электролитов крови (калий, натрий, хлориды).
6. Дополнительная информация, влияющая на течение и исход заболевания
6.1 Осложнения
Синдром Фанкони, псевдогипоальдостеронизм (тип I), нефрогенный несахарный диабет: тяжелая дегидратация.
Глюкозо-галактозный синдром мальабсорбции (тип B): тяжелая дегидратация, нефрокальциноз.
Дистальный РТА (I тип): нефрокальциноз.
6.2 Исходы и прогноз
6.2.1 Проксимальные тубулопатии
6.2.1.1 Гипофосфатемический рахит
При раннем установлении диагноза гипофосфатемического рахита, терапия фосфатом и 1,25-дигидрокси-витамином D3 способствует излечению рахитоподобных деформаций, структура костной ткани полностью не восстанавливается.
6.2.1.2 Проксимальный ренальный тубулярный ацидоз (II тип)
Длительная подщелачивающая терапия эффективна, а при транзиторном младенческом типе вызывает быстрое увеличение роста и с возрастом может быть прервана без опасности рецидива синдрома.
6.2.1.3 Синдром Фанкони (де Тони-Дебре)
Прогноз зависит от причины, обусловившей заболевание, тяжести почечных и экстраренальных проявлений. Идиопатический синдром Фанкони (де Тони-Дебре) может приводить к хронической почечной недостаточности в подростковом или в зрелом возрасте.
Нефропатический цистиноз ведет к хронической почечной недостаточности, нарушениям зрения, гипотиреозу, прогрессирующим неврологическим расстройствам, миопатии. Раннее назначение специфической терапии цистиноза позволяет отсрочить наступление хронической почечной недостаточности и улучшить физическое развитие.
6.2.1.4 Ренальная глюкозурия
При изолированной ренальной глюкозурии прогноз благоприятен. При глюкозо-галактозном синдроме мальабсорбции прогноз благоприятен, если лечение начато до появления нефрокальциноза.
6.2.2 Петлевые тубулопатии
6.2.2.1 Синдром Барттера
В большинстве случаев прогноз благоприятный. Интеллектуальное развитие не страдает. При неонатальном синдроме Барттера гиперкальциурия сохраняется, нефрокальциноз медленно прогрессирует, приводя к почечной недостаточности.
6.2.3 Дистальные тубулопатии
6.2.3.1 Синдром Гительмана
Прогноз при синдроме Гительмана в большинстве случаев благоприятный.
6.2.3.2 Дистальный ренальный тубулярный ацидоз (I тип)
При дистальном РТА прогноз благоприятен, если лечение начато до появления нефрокальциноза. Нефрокальциноз медленно прогрессирует, приводя к почечной недостаточности.
6.2.3.3 Псевдогипоальдостеронизм
При аутосомно-рецессивном варианте псевдогипоальдостеронизма тип I (полиорганная форма) возможны летальные исходы. При аутосомно-доминантном варианте (ренальная форма) прогноз более благоприятен.
6.2.3.4 Нефрогенный несахарный диабет
Излечения от врожденного нефрогенного несахарного диабета не наступает. В грудном возрасте возможен летальный исход на фоне злокачественной гипертермии, не поддающейся лечению антипиретиками, вследствие быстро развившейся дегидратации. Со 2-го года жизни прогноз улучшается в связи с появлением жажды и способности адекватного приема жидкости.
6.2.3.5 Синдром Лиддла
Несвоевременная диагностика синдрома Лиддла и отсутствие адекватной терапии тяжелой артериальной гипертензии может привести к развитию хронической почечной недостаточности, вплоть до терминальной стадии.
Критерии оценки качества медицинской помощи
Вид медицинской помощи
Специализированная медицинская помощь
Условия оказания медицинской помощи
Стационарно / в дневном стационаре
Форма оказания медицинской помощи
Критерий
Выполнено исследование кислотно-щелочного состояния крови
Выполнено исследование биохимического анализа крови (калий, натрий, хлориды, кальций, фосфор, креатинин, глюкоза)
Выполнено исследование общего анализа мочи (глюкоза, белок)
Выполнено исследование биохимического анализа мочи (глюкоза, фосфаты, кальций, белок)
Выполнено исследование рН свежевыпущенной мочи
Выполнено УЗИ почек
Выполнено измерение артериального давления
Список литературы
Приложение А1. Состав рабочей группы
Баранов А.А. академик РАН, профессор, д.м.н., Председатель Исполкома Союза педиатров России. Награды: Орден Трудового Красного Знамени, Орден Почета, Орден «За заслуги перед Отечеством» IV степени, Орден «За заслуги перед Отечеством» III степени
Намазова-Баранова Л.С. академик РАН, профессор, д.м.н., заместитель Председателя Исполкома Союза педиатров России..
Цыгин А.Н., профессор, д.м.н., член Союза педиатров России
Сергеева Т.В., профессор, д.м.н., член Союза педиатров России
Чумакова О.В., профессор, д.м.н., член Союза педиатров России
Паунова С.С., профессор, д.м.н.
Зокиров Н.З., профессор, д.м.н., член Союза педиатров России
Комарова О.В., д.м.н., член Союза педиатров России
Таточенко В.К., профессор, д.м.н., член Союза педиатров России
Бакрадзе М.Д., профессор, д.м.н., член Союза педиатров России
проф., д.м.н. Цыгина Е.Н., член Союза педиатров России
Зробок О.А., к.м.н., член Союза педиатров России
Вашурина Т.В., к.м.н., член Союза педиатров России
Маргиева Т.В., к.м.н., член Союза педиатров России
Лупан И.Н., к.м.н., член Союза педиатров России
Каган М.Ю., к.м.н., член Союза педиатров России
Авторы подтверждают отсутствие финансовой поддержки/конфликта интересов, который необходимо обнародовать.
Приложение А2. Методология разработки клинических рекомендаций
Целевая аудитория данных клинических рекомендаций:
Методы, используемые для сбора/селекции доказательств: поиск в электронных базах данных.
Методы, использованные для оценки качества и силы доказательств:
Методы, использованные для анализа доказательств:
Описание методов, использованных для анализа доказательств
При отборе публикаций, как потенциальных источников доказательств, использованная в каждом исследовании методология изучается для того, чтобы убедиться в ее валидности. Результат изучения влияет на уровень доказательств, присваиваемый публикации, что в свою очередь, влияет на силу рекомендаций.
Для минимизации потенциальных ошибок каждое исследование оценивалось независимо. Любые различия в оценках обсуждались всей группой авторов в полном составе. При невозможности достижения консенсуса привлекался независимый эксперт.
Методы, использованные для формулирования рекомендаций: консенсус экспертов.
Индикаторы доброкачественной практики (Good Practice Points – GPPs)
Рекомендуемая доброкачественная практика базируется на клиническом опыте авторов разработанных рекомендаций.
Экономический анализ
Анализ стоимости не проводился и публикации по фармакоэкономике не анализировались.
Метод валидации рекомендаций
Описание метода валидации рекомендаций
Настоящие рекомендации в предварительной версии были рецензированы независимыми экспертами, которых, прежде всего, попросили прокомментировать, насколько доступна для понимания интерпретация доказательств, лежащая в основе рекомендаций.
От врачей первичного звена (аллергологов-иммунологов) получены комментарии в отношении доходчивости изложения данных рекомендаций, а также их оценка важности предлагаемых рекомендаций, как инструмента повседневной практики.
Все комментарии, полученные от экспертов, тщательно систематизировались и обсуждались членами рабочей группы (авторами рекомендаций). Каждый пункт обсуждался в отдельности.
Консультация и экспертная оценка
Проект рекомендаций был рецензирован независимыми экспертами, которых, прежде всего, попросили прокомментировать доходчивость и точность интерпретации доказательной базы, лежащей в основе рекомендаций.
Рабочая группа
Для окончательной редакции и контроля качества рекомендации были повторно проанализированы членами рабочей группы, которые пришли к заключению, что все замечания и комментарии экспертов приняты во внимание, риск систематических ошибок при разработке рекомендаций сведен к минимуму.
Основные рекомендации
Сила рекомендаций на основании соответствующих уровней доказательств приводятся при изложении текста рекомендаций (уровни достоверности и сила рекомендаций сформулированы на основании проведенного анализа – табл. 1-2).
Уровень
Тип данных
Метаанализ рандомизированных контролируемых исследований (РКИ)
Данные получены по результатам одного РКИ
Доказательства получены на основе метаанализов исследовании? без рандомизации
Хотя бы одно хорошо выполненное квазиэкспериментальное исследование
Хорошо выполненные неэкспериментальные исследования: сравнительные, корреляционные или «случай-контроль»
Экспертное консенсусное мнение либо клинический опыт признанного авторитета
Большие двойные слепые плацебоконтролируемые исследования, а также данные, полученные при мета-анализе нескольких РКИ
Реальный эффект соответствует предполагаемому
Небольшие рандомизированные и контролируемые исследования, при которых статистические данные построены на небольшом числе больных.
Истинный эффект близок к предполагаемому, но есть вероятность различий
Нерандомизированные клинические исследования на ограниченном количестве пациентов
Истинный эффект может значительно отличаться от предполагаемого
Выработка группой экспертов консенсуса по определённой проблеме
Предполагаемый эффект очень неопределённый и в частом проценте случаев может быть далёк от истины
Приложение А3. Связанные документы
Порядки оказания медицинской помощи:
Стандарты оказания медицинской помощи:
Приложение В. Информация для пациентов
Тубулопатии – канальцевые болезни почек, характеризуемые различными нарушениями тубулярного транспорта электролитов, минералов, воды и органических субстанций, наследственного (первичные тубулопатии) или приобретенного характера (вторичные тубулопатии).
Частота встречаемости среди детского населения крайне редка, в связи с чем четкая топическая диагностика канальцевых нарушений представляет определенные трудности.
Несмотря на разнообразие тубулопатий, основу их диагностики составляет раннее выявление таких клинических симптомов, как артериальная гипертензия/гипотензия, полидипсия, полиурия, рахитоподобные изменения, с последующим исследованием и определением нарушений кислотно-щелочного состояния (метаболический ацидоз/алкалоз), натрий-калиевого и кальций-фосфорного гомеостаза
Быстрое назначение адекватной патогенетической терапии позволяет предотвратить многие серьезные осложнения.
Диагностика и лечение пациентов с тубулопатиями проводится врачом нефрологом.
Приложение Г.
… ж – лекарственный препарат, входящий в Перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения на 2016 год (Распоряжение Правительства РФ от 26.12.2015 N 2724-р)
… вк – лекарственный препарат, входящий в Перечень лекарственных препаратов для медицинского применения, в том числе лекарственных препаратов для медицинского применения, назначаемых по решению врачебных комиссий медицинских организаций (Распоряжение Правительства РФ от 26.12.2015 N 2724-р)