Наследственные аминоацидопатии что это
Скрининговое обследование для исключения врождённых (наследственных) «ошибок» метаболизма по типу аминоацидопатий и нарушений бета-окисления жирных кислот. Показано новорождённым в первые недели жизни после начала грудного или искусственного вскармливания.
Аминокислоты
1. Аланин (Ala)
2. Аргинин (Arg)
3. Аспарагиновая кислота (Asp)
4. Валин (Val)
5. Глицин (Gly)
6. Глутаминовая кислота (Glu)
7. Лейцин (Leu)
8. Метионин (Met)
9. Орнитин (Orn)
10. Пролин (Pro)
11. Тирозин (Tyr)
12. Фенилаланин (Phe)
13. Цитруллин (Cit)
14. Свободный карнитин (C0)
15. Ацетилкарнитин (C2)
16. Пропионилкарнитин (C3)
17. Бутирилкарнитин (C4)
18. Изовалерилкарнитин (C5)
19. Глутарилкарнитин (C5DC)
20. Гексаноилкарнитин (C6)
21. Октаноилкарнитин (C8)
22. Деканоилкарнитин (C10)
23. Додеканоилкарнитин (C12)
24. Тетрадеканоилкарнитин (C14)
25. Гексадеканоилкарнитин (C16)
26. Стеароилкарнитин (C18)
Скрининг новорожденных для исключения наследственных «ошибок» метаболизма по типу аминоацидопатий.
Высокоэффективная жидкостная хроматография с тандемным масс-спектрометрическим детектированием (ВЭЖХ-МС/МС).
Мкмоль/л (микромоль на литр).
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
Общая информация об исследовании
Скрининг позволяет исключить нижеперечисленные аминоацидопатии:
− болезнь с запахом кленового сиропа мочи (лейциноз);
− цитруллинемия типа 1, неонатальная цитруллинемия;
− аргининосукциновая ацидурия (АСА)/ недостаточность аргининосукцинатлиазы;
− тирозинемия типа 1;
− тирозинемия типа 2;
− гомоцистинурия/недостаточность цистатионин бета-синтетазы;
Когда назначается исследование?
Показания к обследованию на наследственные нарушения метаболизма аминокислот в раннем возрасте:
− сходные случаи заболевания в семье;
− случаи внезапной смерти ребенка в раннем возрасте в семье;
− резкое ухудшение состояния ребенка после кратковременного периода нормального развития (бессимптомный промежуток может составлять от нескольких часов до нескольких недель);
− необычный запах тела и/или мочи («сладкий», «мышиный», «вареной капусты», «потных ног» и др.);
− нарушения ритма дыхания (брадипноэ, тахипноэ, апноэ);
− нарушения со стороны других органов и систем (поражение печени, гепатоспленомегалия, кардиомиопатия, ретинопатия);
Что означают результаты?
Возраст
Аминокислоты
Аспарагиновая кислота (Asp)
Глутаминовая кислота (Glu)
Ацилкарнитины
Свободный карнитин (C0)
Аспарагиновая кислота (Asp)
Глутаминовая кислота (Glu)
Свободный карнитин (C0)
Кто назначает исследование?
Неонатолог, педиатр, невропатолог, инфекционист.
5 Общий анализ мочи с микроскопией
12 Клинический анализ крови: общий анализ, лейкоцитарная формула, СОЭ (с микроскопией мазка крови при выявлении патологических изменений)
164 Анализ крови на органические кислоты
92 Анализ мочи на органические кислоты
18 Метилентетрагидрофолатредуктаза (MTHFR). Выявление мутации A1298C (Glu429Ala)
18 Метилентетрагидрофолатредуктаза (MTHFR). Выявление мутации C677T (Ala222Val)
18 Метионинсинтаза (MTR). Выявление мутации A2756G (Asp919Gly)
18 Метионин-синтаза-редуктаза (MTRR). Выявление мутации A66G (Ile22Met)
Особенности клинических проявлений и лечения отдельных нозологических форм наследственных болезней обмена веществ

Рассмотрены основные клинические подходы к диагностике и терапии основных жизнеугрожающих наследственных заболеваний обмена аминокислот, относящихся к группе органических ацидурий, таких как болезнь кленового сиропа мочи (БКСМ). Описаны клинико-генетическ
Резюме. Рассмотрены основные клинические подходы к диагностике и терапии основных жизнеугрожающих наследственных заболеваний обмена аминокислот, относящихся к группе органических ацидурий, таких как болезнь кленового сиропа мочи (БКСМ). Описаны выделяют клинико-генетические формы БКСМ и особенности ее клинических проявлений, а также методы диагностики и лечения.
Указывается, что классический вариант БКСМ является одним из самых изменчивых и опасных наследственных метаболических состояний: резкие подъемы уровня лейцина и α-кетоизокапроновой кислоты вызывают метаболическую энцефалопатию и критический отек мозга, в то время как хронический дисбаланс аминокислот и нейротрансмиттеров представляет опасность в плане снижения интеллектуальных функций, приводит к снижению памяти и внимания, умственной нетрудоспособности, нарушению исполнительных функций и развитию психического заболевания. Строгая диетическая терапия с ограничением патогенетически значимых аминокислот и назначением специализированных аминокислотных смесей диетического (лечебного) питания является основным методом лечения больных, назначается при подозрении на наследственное нарушение аминокислотного обмена сразу после взятия анализов. Препарат лечебного питания должен замещать около 50% суточной потребности пациента в белке.
Наличие возможностей ранней диагностики и своевременно начатой диетотерапии с применением специализированных продуктов лечебного (диетического) питания больных с органическими ацидуриями позволяет повысить выживаемость больных, снизить тяжесть когнитивных и психических нарушений, а также частоту возникновения метаболических кризов и выраженность неврологических нарушений.
Своевременная диагностика и лечение наследственных болезней обмена веществ представляют собой одну из важных задач в практике неонатолога, педиатра, невролога. В связи с внедрением в нашей стране обязательного неонатального скрининга — массового обследования новорожденных детей на наиболее распространенные врожденные и наследственные заболевания, многие проблемы оказались решены, однако для ряда других редких жизнеугрожающих наследственных заболеваний обмена аминокислот, относящихся к группе органических ацидурий, проблемы ранней диагностики и своевременного лечения до сих пор остаются нерешенными [1].
Наибольшую группу риска представляют наследственные болезни аминокислотного обмена (НБАО) и органические ацидурии. В настоящее время известно, что органические ацидурии (ацидемии) занимают значительную долю среди редких заболеваний по степени высокого риска смертности среди тяжелобольных детей. Еще в 1954 г. доктор Джон Менкес и его коллеги описали семейный случай, когда у четырех детей (братьев и сестер), рожденных здоровыми, в течение первой недели жизни отмечались признаки прогрессирующей энцефалопатии и запаха мочи, «поразительно похожего на кленовый сироп», с дальнейшим развитием летального исхода в возрасте трех месяцев с клинической картиной отека мозга [2].
Среди группы редких наследственных заболеваний обмена аминокислот, относящихся к группе органических ацидурий (ацидемий), выделяют несколько наиболее частых жизнеугрожающих нарушений: болезнь «кленового сиропа» у детей — МКБ-10: E71.0, метилмалоновая ацидурия у детей — МКБ-10: Е71.1, пропионовая ацидурия (ацидемия) у детей — МКБ-10: Е71 [3–5]. Особенностью классических форм заболеваний этой группы является высокая степень летальности в первые дни и недели жизни детей ввиду полиморфизма клинических проявлений, отсутствия своевременно поставленного диагноза и начала патогенетической диетотерапии.
В статье разбираются основные клинические подходы к диагностике и терапии основных жизнеугрожающих наследственных заболеваний обмена аминокислот, относящихся к группе органических ацидурий.
Методы исследования
Нозологии
Органические ацидурии (ацидемии) представляют собой класс наследственных метаболических нарушений, возникающих вследствие дефектов промежуточного метаболического пути окисления аминокислот, углеводов и жирных кислот.
Классические формы органических ацидурий (ацидемий) у новорожденных первой недели жизни имеют сходную клиническую картину и могут проявляться нарастающей тяжелой клинической неврологической симптоматикой (генерализованными судорогами, вялостью, общим угнетением функций центральной нервной системы, которая чередуется с повышенной возбудимостью, отказом от пищи, упорной рвотой, мышечным гипертонусом, признаками обезвоживания) и при отсутствии своевременно постановленного диагноза и лечения зачастую заканчиваются летально до того, как становится очевидной специфическая метаболическая картина наследственного заболевания. При невыраженных дефектах и относительно благоприятном периоде новорожденности, в более старших возрастных группах пациенты длительно наблюдаются у педиатра или невролога под маской самых различных заболеваний: детского церебрального паралича, различных вариантов умственной отсталости, эпилепсии, резистентной к антиконвульсантам, и метаболических кризов неясной этиологии.
Болезнь кленового сиропа мочи (БКСМ, или MSUD от англ. Maple Syrup Urine Disease) относится к группе редких генетических заболеваний и обусловлена врожденным дефицитом фермента дегидрогеназы кетокислот с разветвленной цепью, необходимой для расщепления (метаболизма) в организме трех аминокислот с разветвленной цепью (АРЦ): лейцина, изолейцина, валина, а также накоплением в биологических жидкостях их дериватов — 2-кетоизокапроновой, 2-кето-3-метилвалериановой, 2-кетоизовалериановой кислот [3].
АРЦ — лейцин, изолейцин и валин являются тремя из девяти незаменимых аминокислот и составляют 35–40% незаменимых аминокислот в составе белков организма и 14% всех аминокислот в скелетных мышцах. Они имеют общие мембранные транспортные системы и ферменты для их переаминирования и необратимого окисления. Они могут быть глюкогенными (валин), кетогенными (лейцин и изолейцин) или теми и другими (изолейцин), поскольку их конечные продукты, сукцинил-КоА и/или ацетил-КоА, могут войти в цикл Кребса для выработки энергии и глюконеогенеза или выступать в качестве прекурсоров для липогенеза и продукции кетоновых тел через ацетил-КоА и ацетоацетат [6].
Мультиферментный комплекс дегидрогеназы кетокислот с разветвленной цепью (BCKD, от англ. branched-chain α-ketoaciddehydrogenase) имеет сложное строение и состоит из 4 белковых соединений: Е1 (α- и β-субъединицы), Е2 и Е3. Коферментом Е1 β-субъединицы служит тиамин пирофосфат. В настоящее время установлена локализация генов, кодирующих отдельные компоненты данного энзимного комплекса: Е1 α-субъединицы (ген BCKDHA) — 19q13.1-q13.2; Е1 β-субъединицы (ген BCKDHB) — 6p22-p21; Е2-протеина (ген DBT) — 1p31; Е3-протеина (ген DLD) — 7q31-q32. Тип наследования — аутосомно-рецессивный. Частота заболевания среди новорожденных в Европе составляет 1:185 000–1:200 000. В России точная частота данного заболевания не определена [3].
Результатом метаболических нарушений при БКСМ является чрезмерное накопление в организме трех АРЦ (лейцина, изолейцина, валина) и их побочных продуктов (2-кетоизокапроновой, 2-кето-3-метилвалериановой, 2-кетоизовалериановой кислот), обладающих общетоксическим, нейротоксическим действием в высоких концентрациях [6].
Высокие уровни лейцина в плазме приводят к конкурентному ингибированию транспорта других крупных нейтральных аминокислот (тирозин, фенилаланин, триптофан, изолейцин, валин, гистидин, метионин, глутамин и треонин) через гематоэнцефалический барьер благодаря их общему транспортеру (LAT1). Уменьшение уровня этих незаменимых аминокислот приводит к снижению синтеза белков и нейротрансмиттеров, таких как дофамин, серотонин, гистамин и S-AdoMet, за счет ограничения доступных предшественников. Кроме того, дефицит кетокислотной дегидрогеназы с разветвленной цепью способствует снижению выработки кетоновых тел из лейцина, которые необходимы для синтеза миелина. В сочетании с нарушением синтеза белка это приводит к тяжелой дисмиелинизации [6–8].
Помимо основного нейротоксического эффекта, обусловленного высоким уровнем патогенетически значимых АРЦ (преимущественно лейцина и его метаболитов), у больных с БКСМ вследствие дефицита нейтральных аминокислот (аланина, глицина, глутамина, тирозина, триптофана и др.) и нарушения их внутриклеточного транспорта отмечается развитие различных нейромедиаторных расстройств.
В патогенезе прогрессирования тяжелых клинических проявлений у больных с БКСМ немаловажную роль также играет кетоацидоз, гипонатриемия, отек и атрофия ткани мозга, вторичная гипераммониемия, недостаточность глюконеогенеза и гипогликемия, а также дисфункция митохондриальной дыхательной цепи и окислительного фосфорилирования.
В настоящее время выделяют следующие клинико-генетические формы БКСМ:
Метилмалоновая ацидемия (ацидурия) — генетически гетерогенное наследственное заболевание из группы органических ацидемий, обусловленное блокированием обмена пропионатов на уровне перехода метилмалонил-КоА в сукцинил-КоА и нарушением метаболизма ряда аминокислот (изолейцин, валин, треонин, метионин), жирных кислот с нечетным числом атомов углерода и холестерина.
Классическая форма заболевания обусловлена дефицитом метималонил-КоАмутазы (ген MUT) с полным (mut0) или частичным (mut-) отсутствием активности фермента. Кофактором данного фермента является витамин В12, поэтому дефекты метаболизма данного фермента также сопровождаются метилмалоновой ацидурией — изолированной или в сочетании с гомоцистинурией [4].
При метилмалоновой ацидемии недостаточная активность метилмалонил-КоА-мутазы приводит к значительному накоплению метилмалоновой кислоты, а также метаболитов, полученных из пропионил-КоА, таких как 3-ОН-пропионат, 2-метилцитрат (продукт реакции пропионил-КоА с оксалоацетатом) и пропионилглицин.
Первоначальные исследования были сосредоточены на изучении метилмалоновой кислоты как основного токсина, в то время как последующие исследования указывают на ключевую роль 3-ОН-пропионата и 2-метилцитрата в развитии различных вторичных биохимических изменений, наблюдаемых при ММА, включая лактатацидоз, гиперглицинемию, гипераммонемию и гипогликемию.
Распространенность заболевания среди новорожденных в странах Европы составляет 1:48 000–61 000; половина случаев представлена В12-резистентными формами. В Российской Федерации точная частота заболевания не определена [4].
Пропионовая ацидемия (ацидурия) — генетически гетерогенное наследственное заболевание из группы органических ацидемий, обусловленное дефицитом пропионил-КоА-карбоксилазы, что ведет к блокированию обмена пропионатов на уровне перехода пропионил-КоА в метилмалонил-КоА и нарушению метаболизма ряда аминокислот (изолейцин, валин, треонин, метионин), жирных кислот с нечетным числом атомов углерода и холестерина. Тип наследования патологии — аутосомно-рецессивный [7].
Патогенез пропионовой ацидемии сходен с патогенезом метилмалоновой ацидемии и связан с накоплением производных пропионовой кислоты вследствие блокирования обмена на уровне перехода пропионил-КоА в метилмалонил-КоА.
Предшественниками пропионатов в организме служат аминокислоты изолейцин, валин, треонин и метионин (50% общего количества пропионатов), жирные кислоты с нечетным числом атомов углерода и холестерин (25%); остальная часть пропионатов образуется в кишечнике в результате деятельности эндогенной флоры. Накопление органических кислот (пропионовой, гидроксипропионовой, метиллимонной и др.) ведет к тяжелому метаболическому кетоацидозу, вторичной гипераммониемии, гиперглицинемии, гипогликемии. Повышенный уровень в крови и высокая почечная экскреция пропионилкарнитина обусловливают истощение запасов карнитина и его вторичный дефицит.
Клинические проявления
Особенностью клинических проявлений при НБАО является тот факт, что ребенок рождается внешне здоровым и в зависимости от степени дефекта фермента симптомы могут проявляться как в первые дни и недели после рождения, так и в более отдаленном периоде в результате хронической интоксикации.
При классической форме БКСМ, с остаточной активностью фермента менее 3%, симптомы заболевания появляются в первые дни после рождения ребенка. На фоне кормления грудным молоком или молочной смесью наблюдается резкое ухудшение общего состояния: вялость, которая может сменяться повышенной возбудимостью, возможно возникновение упорной рвоты, отказа от пищи, судорог, коматозного состояния, цианоза, нарушения дыхания (апное).
В случае нарастания признаков энцефалопатии у новорожденных детей с БКСМ в плазме крови отмечаются повышение концентрации патогенетически значимых аминокислот (лейцина, изолейцина, валина), включая аллоизолейцин (allo-ILE), а также общее нарушение соотношения концентраций аминокислот спустя 12–24 ч после рождения. Повышение декарбоксилазы производных α-кетокислоты (BCKAs) и генерализованная кетонурия, раздражительность и снижение аппетита (плохое сосание) могут возникать через 24–72 ч; усугубление энцефалопатии, проявляющееся в виде вялости, перемежающегося апноэ, опистотонуса и стереотипных движений, таких как «фехтование» и «езда на велосипеде», на 4–5 день. При наличии прогрессирующих изменений кома и центральная дыхательная недостаточность могут возникнуть на 7–10 дни. Другие формы заболевания БКСМ с различной степенью частичной активности ферментов, включая промежуточную, интермиттирующую, тиамин-зависимую, могут привести к тяжелой хронической метаболической интоксикации и развитию хронической энцефалопатии с катаболическим стрессом.
У детей старше 1 года при отсутствии своевременно поставленного диагноза прогрессируют признаки задержки психомоторного развития, умственная отсталость, эритематозные высыпания, частые инфекционные заболевания, судорожный синдром. У большинства больных БКСМ отмечается специфический запах мочи.
Схема основных клинических проявлений и метаболических нарушений, наблюдающихся при БКСМ, метилмалоновой/пропионовой ацидуриях, представлена в табл. 1.
_575x.gif)
Диагностика
Классическая диагностика БКСМ проводится на базе медико-генетического центра врачом-генетиком и основана на анализе родословной, данных анамнеза, клинических проявлений, результатах анализа уровня аминокислот лейцина, изолейцина, валина в крови с подсчетом соотношения лейцин/аланин, определении почечной экскреции органических кислот — 2-кетоизокапроновой, 2-кето-3-метилвалериановой, 2-кетоизовалериановой.
На практике, в случае подозрения на заболевание при наличии острого метаболического криза или в случае тяжелого состояния ребенка в стационаре, рекомендуется определение кислотно-щелочного состояния крови (cito!). Клиническая картина у ребенка с БКСМ в тяжелом состоянии сопровождается метаболическим кетоацидозом, гипонатриемией, гипогликемией, угрозой развития отека мозга и требует интенсивной коррекции выявленных нарушений. Для подтверждения диагноза или при подозрении на наследственное нарушение аминокислотного обмена необходима консультация генетика. Исследование содержания аминокислот, органических кислот, ацилкарнитинов в крови и моче проводят до начала лечения [3, 5, 6].
Возможные колебания концентрации аминокислот в плазме крови у больных с БКСМ представлены в табл. 2.
.gif)
Аллоизолейцин является химическим производным изолейцина и представляет собой самый чувствительный и специфический диагностический маркер для всех форм БКСМ. Аллоизолейцин плазмы составляет 2 = 37,9, р 2 = 0,51, р = 0,477). Следует также отметить, что у двадцати детей (49%) заболевание было обнаружено благодаря государственной программе неонатального скрининга [8]. Таким образом, стандартный протокол, включающий методы раннего выявления заболевания, своевременного начала контролируемой диетотерапии больных позволяет значительно снизить смертность и увеличить продолжительность жизни пациентов с БКСМ.
При отсутствии своевременной диагностики, нутритивной коррекции развитие эпизодов метаболического криза на фоне декомпенсации в силу различных причин является потенциально опасным для пациента и затратным для системы здравоохранения. Проведение трансплантации печени позволяет в ряде случаев восстановить гомеостаз АРЦ, однако это имеет непродолжительный эффект [8]. Таким образом, сохраняется необходимость в совершенствовании возможностей своевременной диагностики данных заболеваний, разработке более универсальных методов улучшенной безопасной, болезнь-модифицирующей терапии.
Заключение
При отсутствии своевременной диагностики и нутритивной коррекции развитие эпизодов метаболического криза на фоне декомпенсации в силу различных причин является потенциально опасным для пациента и затратным для системы здравоохранения.
Возможности ранней диагностики и своевременно начатой диетотерапии с применением специализированных продуктов лечебного (диетического) питания больных с органическими ацидуриями позволяют повысить выживаемость больных.
В настоящее время диетотерапия с применением современных, адаптированных по возрасту специализированных продуктов лечебного (диетического) питания является основным методом лечения НБАО, который позволяет контролировать метаболические процессы в приемлемых рамках, снизить тяжесть и выраженность неврологических, когнитивных и психических нарушений, а также частоту возникновения метаболических кризов.
Материал подготовлен при поддержке компании ООО «Нутриция».
Литература/References
Л. В. Горошко 1
Е. Г. Бакулина, доктор медицинских наук
АНМО СКККДЦ, Ставрополь
Особенности клинических проявлений и лечения отдельных нозологических форм наследственных болезней обмена веществ/ Л. В. Горошко, Е. Г. Бакулина
Для цитирования: Лечащий врач № 6/2020; Номера страниц в выпуске: 12-17
Теги: аминокислотный обмен, дети, неврологические нарушения, когнитивные нарушения
Неонатальный скрининг

Неонатальный скрининг, ласково именуемый в нашей стране «пяточка», является одним из первых важных исследований новорожденного. В России скрининг преимущественно направлен на выявление пяти наследственных болезней обмена: фенилкетонурии, врожденного гипотиреоза, врожденной дисфункции коры надпочечников (ВДКН), галактоземии и муковисцидоза. За рубежом этот список расширен до 50 различных заболеваний, в некоторых штатах Америки их свыше 60. Здоровый доношенный новорожденный допускается к скринингу на 4–5 сутки, недоношенный — на седьмой день после рождения. Заболевания, на выявление которых направлен скрининг, никак не проявляют себя в периоде новорожденности, но их ранняя диагностика и своевременно начатое патогенетическое лечение существенно влияют на прогноз и качество жизни ребенка. Помимо исследования крови проводится аудиометрия для оценки слуха и пульсоксиметрия для скрининга пороков сердца, но в данной статье мы преимущественно сосредоточимся на тестировании крови.
Разработка программы скрининга началась в шестидесятых годах прошлого века, когда Роберт Гатри создал технологию тестирования сухих отпечатков крови на фильтровальной бумаге. Первым заболеванием, которое стало кандидатом для массовой диагностики, была фенилкетонурия, так как ее раннее выявление и коррекция питания способны предотвратить развитие тяжелых неврологических нарушений. Затем к скринингу добавилось еще несколько заболеваний: врожденный гипотиреоз, ВДКН, галактоземия и муковисцидоз. Тандемная масс-спектрометрия (ТМС) позволила значительно расширить список заболеваний, добавив к болезням обмена веществ гемоглобинопатии, спинальную мышечную атрофию, тяжелый комбинированный иммунодефицит и др.
Таблица 1 | Рекомендуемая The American College of Obstetricians and Gynecologists (ACOG) скрининг-панель для врожденных заболеваний
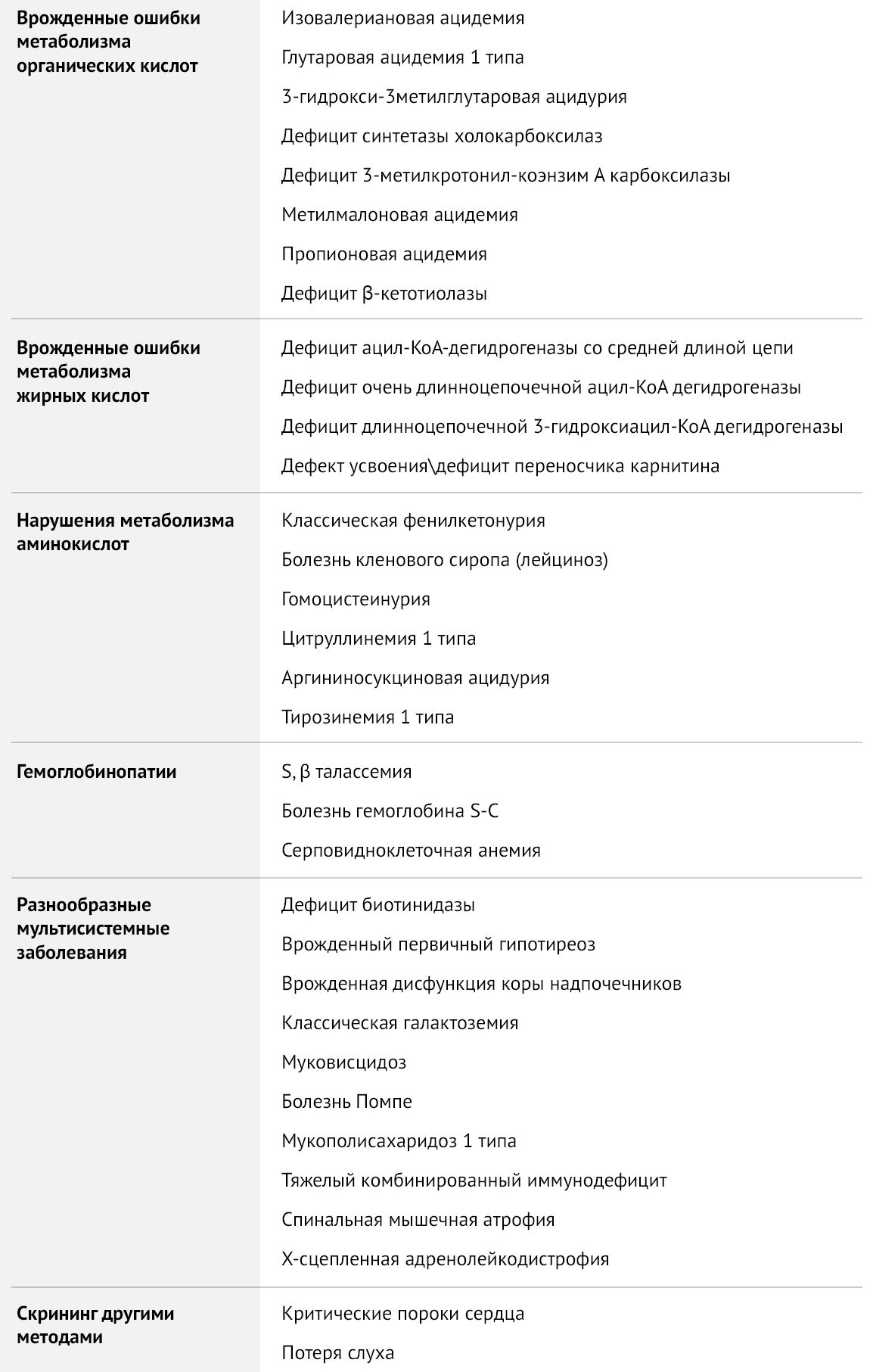
В данной статье будут рассмотрены два скрининга, доступные в нашей стране: обязательный, включающий тестирование на пять заболеваний (врожденный гипотиреоз, ВДКН, фенилкетонурия, галактоземия, муковисцидоз) и расширенный скрининг на наследственные нарушения метаболизма.
Обязательный скрининг
На 4–5 сутки после рождения здорового доношенного ребенка или на седьмые сутки жизни недоношенного ребенка проводится тестирование методом «сухого пятна».
Фенилкетонурия (в современной классификации ― ФАГ-зависимая ФКУ) обусловлена мутацией гена фенилаланингидроксилазы и относится к числу аминокислотных аминоацидопатий. В норме фенилаланин (ФА) путем реакций гидроксилирования превращается в тирозин, однако в случае мутации вышеназванного гена активность превращающего фермента снижается, создается дефицит тирозина одновременно с избытком ФА, образующего токсичные метаболиты (фенилацетат, фенилпируват, фениллактат). Снижение образования тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов, а избыток ФА приводит к дисбалансу аминокислот в тканях мозга, обусловленному торможением их всасывания в желудочно-кишечном тракте или нарушением реабсорбции из почечных канальцев, нарушению образования или стабилизации полирибосом, снижению синтеза миелина, норадреналина и серотонина. Также за счет конкурентного ингибирования создается дефицит тирозиназы, что в совокупности с дефицитом тирозина приводит к снижению образования меланина и гипопигментации.
Основной проблемой пациентов с ФКУ являются нарушения функции ЦНС: от сонливости, вялости, отсутствия аппетита в период манифестации в 2–6 месяцев до тяжелых нарушений психомоторного развития в будущем; нередко развиваются атаксия, гиперкинезы, тремор рук, парезы по центральному типу. Единственный способ предотвратить развитие вышеназванных нарушений — назначение гипофенилаланиновой диеты с момента рождения с поддержанием низкого уровня фенилаланина в течение всей жизни.
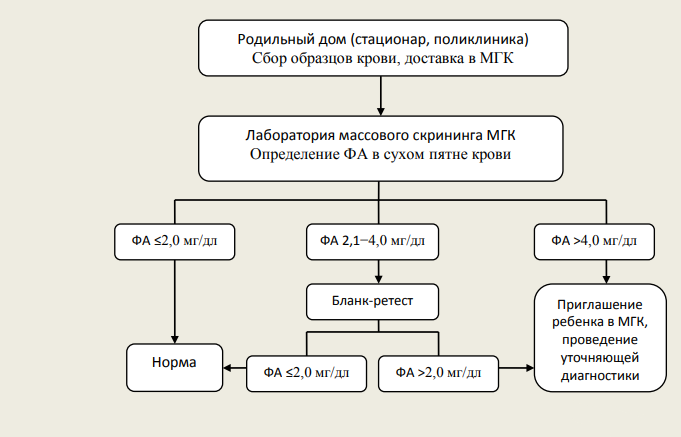
Рисунок 1 | Интерпретация результатов исследования на наличие фенилкетонурии
ВДКН обусловлена дефицитом ферментов и транспортных белков, участвующих в биосинтезе кортизола. Наиболее часто встречается дефицит 21-гидроксилазы, что в свою очередь приводит к дефициту кортизола и альдостерона и ответному увеличению секреции АКТГ и гиперплазии коры надпочечников. В условиях дефицита фермента происходит значительное накопление предшественников гормонов, что приводит к увеличению синтеза тестостерона, не зависящего от 21-гидроксилазы. В итоге у пациента формируется надпочечниковая недостаточность и гиперандрогения. Гормональным маркером дефицита 21-гидроксилазы является уровень 17-гидроксипрогестерона (17-ОНП), определяемый в рамках неонатального скрининга. Результат трактуется как положительный, если при двукратном тестировании образца уровень 17-ОНП у доношенных новорожденных составляет ≥ 20 нг/мл. У недоношенных детей при заборе крови на 7–8 сутки после рождения скрининговый результат трактуется как положительный при следующих уровнях 17-ОНП: на сроке 23–32 недели гестации ― ≥ 65 нг/мл; на сроке 33–36 недель гестации ― ≥ 40 нг/мл.
Врожденный гипотиреоз в большинстве случаев вызван дефектами самой щитовидной железы (первичный гипотиреоз). Причины первичного врожденного гипотиреоза можно в широком смысле классифицировать как неспособность щитовидной железы нормально развиваться (дисгенезия) или неспособность структурно нормальной щитовидной железы производить нормальные количества гормона (дисгормоногенез). Дисгенезия щитовидной железы, охватывающая весь спектр агенеза, гипоплазии и эктопии, является наиболее частой причиной врожденного гипотиреоза. В то время как это заболевание остается наиболее частой причиной врожденного гипотиреоза, частота возникновения дисгормоногенеза за последние несколько десятилетий увеличилась. В то время как на дисгормоногенез приходится только 15 % врожденного гипотиреоза, диагностированного в первые дни скрининга новорожденных, у 30–40 % младенцев, прошедших скрининг по современным протоколам, имеется эктопическая щитовидная железа, соответствующая одной из форм дисгормоногенеза. В отличие от дисгенезии щитовидной железы, при которой моногенная причина присутствует только у небольшого количества пациентов, дисгормоногенез часто возникает из-за генетического дефекта на каком-либо этапе синтеза тиреоидных гормонов.
Учитывая разнообразие функций тиреоидных гормонов в организме человека, врожденный гипотиреоз характеризуется разнообразием клинических проявлений с поражением всех органов и систем. При отсутствии своевременного лечения на первый план выходит задержка психомоторного и речевого развития, затем наступают отставание в физическом развитии и задержка полового развития. Основной задачей скрининга является наиболее раннее выявление детей с подозрением на врожденный гипотиреоз.

Рисунок 2 | Интерпретация результатов исследования на наличие врожденного гипотиреоза
Галактоземия — аутосомно-рецессивное наследственное нарушение обмена углеводов, при котором в организме накапливается избыток галактозы и ее метаболитов. В норме галактоза образуется в результате гидролиза лактозы в кишечнике либо в процессе ферментных реакций, обмена гликопротеинов и гликолипидов. Галактоза является материалом для образования клеточных мембран, нервной ткани, нервных окончаний и т. д. В результате ферментных реакций она превращается в глюкозу, и именно дефицит галактозо-1-фосфатуридилтрансферазы лежит в основе патогенеза данного заболевания. Метаболиты галактозы обладают повреждающим действием. Так, галактитол проникает в хрусталик глаза, приводя к повышению осмотического давления, электролитным нарушениям и денатурации белка с формированием катаракты. Другие метаболиты обладают гепато-, нейро- и нефротоксическим действиями, а также вызывают гемолиз эритроцитов. Тормозящее влияние метаболитов галактозы на углеводный обмен приводит к гипогликемии.

Рисунок 3 | Интерпретация результатов исследования на наличие галактоземии
Муковисцидоз — аутосомно-рецессивное заболевание, связанное с мутацией гена МВТР (трансмембранного регулятора муковисцидоза). МВТР является хлорным каналом, мутации гена которого нарушают не только транспорт, но и секрецию ионов хлора. При затруднении их прохождения через клеточную мембрану увеличивается реабсорбция натрия железистыми клетками, нарушается электрический потенциал просвета, что вызывает изменение электролитного состава и дегидратацию секрета желез внешней секреции. В результате выделяемый секрет становится чрезмерно густым и вязким. Поражаются все экзокринные железы организма: печень, поджелудочная железа, мочеполовая система, но наиболее ярко муковисцидоз проявляет себя со стороны органов дыхания, провоцируя бронхообструкцию, дыхательную и сердечную недостаточность, легочную гипертензию.

Рисунок 4 | Интерпретация результатов исследования на наличие муковисцидоза
Расширенный скрининг
Органические ацидемии — группа аутосомно-рецессивных наследственных заболеваний обмена, в основе патогенеза которых лежит дефицит ферментов, участвующих в метаболизме белков, что приводит к повышению уровня кетоновых тел, обладающих токсическим действием на различные органы и ткани, в частности, на ЦНС. Данные заболевания манифестируют уже в стадии декомпенсации, как правило, в период с первой недели до первого года жизни. Триггерами служат стресс, длительное голодание, инфекционные заболевания, иммунизация, реже — чрезмерное употребление белковой пищи. Проявляются преимущественно неврологической симптоматикой: нарушение сознания вплоть до комы, эпилептические приступы, нарушение мышечного тонуса, у детей старшего возраста — нарушения психоречевого развития, атаксия, очаговые неврологические симптомы, синдром Рейе (острая печеночная недостаточность, сочетающаяся с энцефалопатией), психические расстройства.
Нарушения окисления жирных кислот — врожденный дефект метаболизма из-за нарушения либо митохондриального β-окисления, либо транспорта жирных кислот с использованием карнитинового транспортного пути. Проявления зависят от нарушения метаболизма конкретной кислоты, но все они имеют общие черты и требуют схожей тактики лечения. В периоде новорожденности метаболические нарушения проявляются тяжелой кардиомиопатией, гипокетотической гипогликемией, дисфункцией печени в первые несколько дней или недель жизни, часто заканчиваясь летально. В младенческом и детском возрасте характерны эпизоды летаргии и рвоты, развивается дисфункция печени и гипокетотическая гипогликемия, энцефалопатия, что может привести к внезапной младенческой смерти. У подростков и во взрослом возрасте дебютируют эпизодическим рабдомиолизом, мышечной слабостью, миалгией. Лечение включает отказ от голодания, симптоматическую терапию развившихся осложнений и включение в рацион добавок, если это необходимо.
Аминоацидопатии
Болезнь кленового сиропа (она же лейциноз) — наследственное заболевание, обусловленное дефицитом дегидрогеназы кетокислот с разветвленной цепью и нарушением метаболизма лейцина, изолейцина, валина (аминокислоты с разветвленной цепью, АКЦР). Повышение уровня АКЦР и их метаболитов, в частности, кетокислот, приводит к кетоацидозу, атрофии ткани головного мозга, нарушению окислительного фосфорилирования в дыхательной цепи митохондрий. Избыток лейцина обладает нейротоксическим эффектом, вызывая дисфункцию астроцитов, апоптоз нейронов и блокируя транспорт через гематоэнцефалический барьер аминокислот, важных для синтеза нейротрансмиттеров.
Гомоцистеинурия — наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот, в частности, метионина. Дефицит цистатион-b-синтазы нарушает преобразование метионина в цистеин. Высокий уровень гомоцистеина связан с образованием некротически-дегенеративных участков в почках, селезенке, слизистой оболочке желудка и сосудах, активацией XII фактора свертывания, способствующего тромбообразованию.
Тирозинемия 1 типа — заболевание, обусловленное дефицитом фумарилацетоацетатгидролазы, в результате чего происходит накопление высокотоксичных фумарил- и малеилацетоацетата, обладающих гепатотоксическим и канцерогенным действием. Конечные метаболиты — сукцинилацетон и сукцинилацетоацетат — являются митохондриальными токсинами, тормозящими фосфорилирование и блокирующими цикл Кребса. Накопление токсинов приводит к прогрессирующему заболеванию печени с развитием печеночной недостаточности, цирроза, тубулопатии с формированием ренальной тубулопатии, гипофосфатемического рахита, синдрома Фанкони. Острая тирозинемия сопровождается развитием гипертрофической кардиомиопатии. Кроме того, нарушается путь синтеза порфирина, ингибируется синтез порфобилиногена, что приводит к кризам, проявление которых напоминает порфирию. Все пациенты подвержены высокому риску развития гепатоцеллюлярной карциномы, вторичной по отношению к циррозу. Без своевременного лечения дети погибают в возрасте 10 лет.