Нейрофиброматоз 1 типа
Нейрофиброматоз 1 типа (НФ1, болезнь Реклингхаузена, периферический НФ) – генетическое заболевание, которое встречается примерно у 1 из 2500-3500 новорожденных детей. Это самое распространенное генетическое заболевание среди редких. По данным американской ассоциации CTF каждый день рождается около 120 человек с нейрофиброматозом 1 типа. Это значит, что новый пациент с НФ1 рождается каждые 12 минут.
Диагноз нейрофиброматоз 1 типа может быть поставлен при наличии сочетания ДВУХ И БОЛЕЕ симптомов:

Данная информация носит ознакомительный характер и не может быть использована для самодиагностики, диагноз может поставить только врач!
Указанные признаки могут встречаться в любом сочетании, но ни один из них в отдельности не является достаточным для диагностики НФ1. Также отсутствие какого-либо из признаков (например, пятен цвета «кофе с молоком», которые присутствуют примерно у 99% больных НФ1) не препятствует постановке диагноза.
Также при нейрофиброматозе 1 типа у пациентов встречаются: эпилептические приступы, опухоли центральной нервной системы (ЦНС), вишневые ангиомы (пятна Кэмпбелла де Моргана), ксантогранулемы, низкорослость, раннее половое созревание и другие более редкие проявления, не являющиеся диагностическими, но требующими внимания.
Симптомы НФ1 проявляются у пациентов индивидуально в различные периоды жизни и практически на всем ее протяжении – часть из них формируется в течение первых двух лет жизни, другие – к 5-7 годам, и так далее. Например, симптомы, имеющие кожные проявления, чаще всего проявляются при рождении, в младенческом возрасте или до достижения ребенком возраста 10 лет. Появление пигментных пятен, пятен кофейного цвета и узелков Лиша, как правило, не представляют опасности для здоровья, хотя могут стать причиной психологического дискомфорта. Примерно в 10-15 лет характерным становится появление нейрофибром, образование которых может проходить внутри тела и затрагивать деятельность различных органов. Гормональные изменения в пубертатный период или в период беременности могут спровоцировать увеличение размера нейрофибром.
Нейрофиброматоз не является противопоказанием к прививкам, проведению массажа, нахождению ребенка на солнце, ЛФК. За исключением случаев, когда указанные процедуры являются противопоказанием при наличии сопутствующего заболевания.
Узнать больше о развитии детей с нейрофиброматозом 1 типа вы можете здесь.
Нейрофиброматоз 1 типа что это
Этиология и встречаемость нейрофиброматоза I типа. Нейрофиброматоз I типа (NF1, MIM №162200) — панэтническое аутосомно-доминантное заболевание с симптомами, чаще проявляющимися в коже, глазах, скелете и нервной системе. NF1 вызван мутациями в гене нейрофибромина (NF1). Болезнь встречается с частотой 1 на 3500 человек, что делает ее одним из наиболее частых аутосомно-доминантных заболеваний.
Приблизительно половина пациентов имеет новые мутации; частота мутаций в гене NF1 — одна из самых высоких среди всех генов человека, приблизительно 1 мутация на 10 000 живых новорожденных. Около 80% новых мутаций отцовского происхождения, однако влияние отцовского возраста на частоту мутации не подтверждено.
Патогенез нейрофиброматоза I типа
NF1 — большой ген (350 килобайт, 60 экзонов), кодирующий нейрофибромин, белок, широко экспрессируемый почти во всех тканях, но наиболее обильно в головном и спинном мозге и периферической нервной системе. Считают, что нейрофибромин регулирует несколько внутриклеточных процессов, в том числе активацию ГТФазы Ras, управляя клеточным делением и функционируя как супрессор опухолевого роста.
В гене NF1 описано более 500 мутаций, большинство из которых уникальны для каждой семьи. Клинические проявления связаны с потерей функции продукта гена; 80% мутаций вызывает укорочение молекулы белка. Мутация как причина болезни может быть выявлена более чем у 95% больных нейрофиброматозом I типа.
Нейрофиброматоз I типа характеризуется крайней клинической изменчивостью, как меж-, так внутрисемейной. Эта изменчивость, вероятно, вызвана комбинацией генетических, негенетических и случайных факторов. Отчетливой корреляции генотип-фенотип не обнаружено, хотя большие делеции чаще наблюдают у больных с неврологическими проблемами.
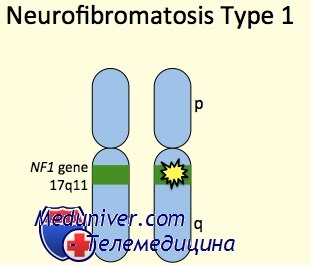
Фенотип и развитие нейрофиброматоза I типа
Нейрофиброматоз I типа — мультисистемное заболевание с неврологическими, мышечно-скелетными, глазными и кожными аномалиями, а также предрасположенностью к новообразованиям. Диагноз нейрофиброматоз I типа может быть поставлен, если обнаружены два или более следующих критериев: шесть или более пятен цвета «кофе с молоком», имеющие в диаметре, по крайней мере, 5 мм (до пубертата) или 15 мм (после пубертата); две или больше нейрофибромы любого типа или одну плексиформную нейрофиброму; веснушки в подмышечной или паховой области; глиому зрительного нерва; два или более узелков Лиша; характерный костный фенотип (сфеноидная дисплазия и уменьшение коры длинных трубчатых костей как с, так и без псевдоартрозов); наличие родственника первой степени родства с нейрофиброматозом I типа.
Почти все больные с нейрофиброматозом I типа без семейного анамнеза не имеют клинических критериев ранее 8 лет. Детям, унаследовавшим нейрофиброматоз I типа, диагноз обычно может быть установлен клинически уже на первом году жизни, так как для них требуется наличие только одного симптома болезни.
Хотя пенетрантность по существу полная, проявления чрезвычайно вариабельны. Множество пятен цвета «кофе с молоком» присутствуют почти у всех больных, веснушки наблюдают в 90% случаев. Множество больных с нейрофиброматозом I типа имеют только кожные симптомы болезни и узелки Лиша на радужке.
Многочисленные нейрофибромы обычно присутствуют у взрослых. Плексиформные нейрофибромы встречаются реже. Глазные проявления включают глиомы зрительного нерва (могут вести к слепоте) и узелки Лиша на радужке. Наиболее тяжелые осложнения со стороны костной системы — сколиоз, дисплазия позвонков, псевдоартрозы и избыточный рост. Часто встречаются также стеноз легочных, почечных и церебральных сосудов и гипертония. Наиболее частые опухоли у детей с нейрофиброматозом I типа (кроме нейрофибром) — глиомы зрительного нерва, опухоли мозга и злокачественные миелоидные болезни.
До половины всех детей с нейрофиброматозом I типа имеют когнитивные проблемы или дефицит внимания, сохраняющиеся во взрослом возрасте.
У больных с симптомами нейрофиброматоза I типа, ограниченными одной областью тела, имеющих здоровых родителей, может диагностироваться сегментный (или локальный) нейрофиброматоз I типа. Сегментный нейрофиброматоз I типа может представлять случайное необычное распределение клинических симптомов или соматический мозаицизм по мутации в гене NF1.
Особенности фенотипических проявлений нейрофиброматоза I типа:
• Возраст начала: от пренатального до позднего детства
• Пятна «кофе с молоком»
• Веснушки в подмышечных и паховых областях
• Нейрофибромы
• Узелки Лиша (гамартомы радужки)
• Плексиформные нейрофибромы
• Глиомы зрительного нерва
• Специфические повреждения костей
Лечение нейрофиброматоза I типа
Нейрофиброматоз I типа — клинический диагноз. Идентификацию мутаций в настоящее время стандартно не проводят из-за большого размера гена и крайней аллельной гетерогенности.
Эффективного лечения нет, следовательно, помощь сфокусирована на симптоматической терапии. Постоянное наблюдение пациентов с нейрофиброматозом I типа должно включать ежегодный медицинский осмотр, проводимый знакомым с этим заболеванием врачом, ежегодный осмотр окулиста в детстве (реже у взрослых), регулярную оценку психомоторного развития в детстве и регулярный контроль артериального давления.
Деформации, вызываемые нейрофиброматозом I типа, — наиболее беспокоящее проявление болезни. Отдельные кожные и подкожные нейрофибромы можно удалить хирургическим способом, если они доставляют косметические или другие неудобства. Плексиформные нейрофибромы, вызывающие обезображивание или беспокойства, также могут быть удалены хирургически. Тем не менее хирургическое вмешательство для этих неоплазм может быть проблематичным, так как они часто интимно спаяны с нервами и имеют тенденцию рецидивировать в месте резекции.
Риски наследования нейрофиброматоза I типа
Пациенты с нейрофиброматозом I типа имеют 50% риска родить ребенка, пораженного болезнью, хотя симптоматика у ребенка может отличаться от имеющейся в семье. Пренатальная диагностика доступна в семьях с известной мутацией в гене NF1, вызывающей болезнь, или информативных по сцеплению. Хотя пренатальный диагноз точен, он не обеспечивает прогностическую информацию из-за крайней фенотипической изменчивости болезни.
Родители больного ребенка, не имеющие никаких признаков болезни, имеют небольшое повышение риска повторения при следующей беременности из-за возможности полового мозаицизма, подтвержденного при нейрофиброматозе I типа.
Пример нейрофиброматоза I типа. Л.М., 2-летний мальчик, проходит обследование в связи обнаружением пяти пятен цвета «кофе с молоком», три из них более 5 мм в диаметре. У него не найдено веснушек в подмышечных или паховых областях, пороков развития кости и нейрофибром. Клинический осмотр обоих родителей не выявил признаков нейрофиброматоза. Генетик сообщил родителям и направившему педиатру, что не обнаружил клинических критериев для нейрофиброматоза I типа.
Ребенок повторно обратился в клинику генетики в 5 лет. У него появились узелки Лиша на радужке глаз и 12 пятен цвета «кофе с молоком», 8 из которых превышали 5 мм в диаметре. Обнаружена также веснушчатость в подмышечных областях с двух сторон. Установлен диагноз нейрофиброматоз I типа; родителям сообщили, что у ребенка новая мутация, следовательно, риск повторения низкий, но не исключен гонадный мозаицизм.
Родители отказались как от молекулярных анализов, так и от пренатальной диагностики при последующей беременности.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Нейрофиброматозы
Нейрофиброматозы – наследственные заболевания, характеризующиеся образованием доброкачественных опухолей в коже, мягких тканях, нервной системе и внутренних органах. Выделяют 6 типов нейрофиброматозов, клинически значимы типы I и II. Общие симптомы включают нейрофибромы на коже, опухоли спинномозговых корешков, слуховых и зрительных нервов, пигментные пятна, костные деформации. Диагностика основана на данных осмотра пациентов, выявлении опухолей с помощью МРТ и КТ спинного и головного мозга, внутренних органов. Лечение симптоматическое – проводится резекция опухолей, рентгенотерапия, химиотерапия.
МКБ-10
Общие сведения
Нейрофибромы – доброкачественные опухоли, развивающиеся из оболочек нервных волокон. Чаще всего располагаются в слоях кожи и подкожной клетчатке, иногда поражают головной мозг, нервные волокна, корешки спинного мозга, мягкие ткани, внутренние органы. Нейрофиброматоз – болезнь, при которой образуются многочисленные нейрофибромы. Распространенность разных типов патологии значительно колеблется: заболеваемость 1 типом составляет 1:2 500, 2 типом – 1:50 000. Другие варианты встречаются еще реже, их точная эпидемиология не определена. Гендерной и расовой предрасположенности не выявлено. Дебют клинических проявлений возможен в любом возрасте, зависит от типа болезни.
Причины нейрофиброматозов
Образование множественных нейрофибром детерминировано генетически. При нейрофиброматозе I существует мутация гена НФ1, расположенного на длинном плече 17 хромосомы. Он относится к генам-супрессорам роста опухолевых тканей, большая часть из которых – нейроэктодермального генеза. При дефекте в гене НФ1 нарушается синтез белков, ответственных за клеточную пролиферацию. Мутации носят характер транслокаций, делеций, инверсий, точковых изменений. Больше 80% из них приводят к синтезу нефункциональных белков или к полному отсутствию белковых молекул. Наследование происходит по аутосомно-доминантному механизму с высокой степенью пенетрации: при наличии мутационного гена у одного из родителей вероятность болезни у ребенка составляет 50%, если оба родителя имеют мутацию, риск повышается до 80-90%. Известны случаи спонтанных мутаций.
Причиной нейрофиброматоза II является мутационное изменение гена НФ2, локализованного на 22 хромосоме. Он кодирует производство белка мерлина (шванномина) – супрессора опухолевого роста. Тип наследования – аутосомно-доминантный с небольшой степенью пенетрации. Передача одного мутантного гена зачастую не проявляется, поскольку второй ген обеспечивает синтез достаточного количества белков. Если он повреждается, синтез нормальных фракций мерлина прекращается, пролиферация клеток усиливается, развивается новообразование. При других типах нейрофиброматозов также существуют мутации в генах, обеспечивающих воспроизведение молекул белков-супрессоров роста опухолей.
Патогенез
Общим патогенетическим механизмом развития нейрофиброматозов является наследственно обусловленная недостаточность какого-либо белка, подавляющего процессы опухолевой пролиферации клеток в тканях нейроэктодермального происхождения. При мутации одного гена производство опухолевых супрессоров прекращается наполовину, равновесие роста и гибели клеток смещается в сторону митотического деления. Нормальный аллельный ген частично компенсирует дефицит белка. Тяжесть нейрофиброматоза определяется тем, насколько дефектный ген влияет на активность белка-супрессора – частично или полностью нарушает функциональность, полностью блокирует производство. Кроме этого, выраженность клинических признаков зависит от сохранности противоопухолевого иммунитета.
Во многих органах и тканях пациентов формируются доброкачественные опухолевые образования, состоящие из соединительной ткани и пигментных клеток. На нервных стволах образуются невриномы; на поверхности кожи – пигментированные области, жировые бляшки, расширенные сосуды; на сетчатке глаз – факоматоз. Изменяется строение костей, они остаются недоразвитыми либо чрезмерно утолщаются, искривляется позвоночный столб.
Симптомы нейрофиброматозов
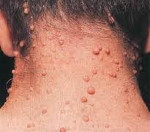
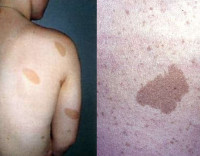
Заболевания проявляются признаками поражения кожи, нервной системы. Классическим клиническим вариантом является нейрофиброматоз типа I, на долю которого приходится 90% случаев болезни. Характерный симптом – гиперпигментация. У больных при рождении или в раннем детстве появляются кожные пятна, цвет которых варьируется от светло-золотистого до коричневого «кофе с молоком». В отдельных случаях пятна имеют фиолетовый или синий оттенок. На радужке глаза обнаруживаются узелки Лиша (пятна пигмента – гамартомы) небольшого размера, белесоватые или светло-бежевые, заметные только при офтальмологическом осмотре. Являются специфическим признаком нейрофиброматоза 1, образуются по мере взросления: у детей до 4 лет распространенность составляет 22%, с 5 до 9 лет – 41%, с 10 до 19 лет – 85%, после 20 лет – 95%.
В период пубертата и позже формируются кожные и плексиформные нейрофибромы, располагающиеся соответственно подкожно (на мелких нервных волокнах, иннервирующих кожу) и на крупных нервах. Они представляют собой небольшие доброкачественные новообразования. Кожные нейрофибромы воспринимаются как косметический дефект, при определенном расположении травмируются. Плексиформные опухоли, локализующиеся по ходу периферических нервов, выявляются на конъюнктиве, веках, в брюшной полости и средостении. Проявляются хронической болью, онемением, судорогами, параличом. Опухоли ЦНС находятся внутри черепа, представлены глиомами зрительных нервных волокон, астроцитомами, эпендимомами, невриномами слухового нерва, менингиомами и нейрофибромами. Клиническая картина определяется размерами новообразований, вовлеченностью мозговых структур в патологический процесс. В детском возрасте диагностируются расстройства психического развития: снижение когнитивных способностей, гиперактивность, редко – деменция.
При тяжелом нейрофиброматозе деформируется костная система. У больных возникает сколиоз, краевые структурные изменения тел позвонков и их отростков, эрозийные поражения краев межпозвоночных отверстий и задних ребер. Характерна атрофия либо, наоборот, гипертрофия трубчатых костей. Кости часто искривлены, на поверхности обнаруживаются периостальные гребни и наслоения. В полостях костей могут образовываться нейрофибромы. Если в процесс вовлекаются кости черепа, появляется внешняя асимметрия, наиболее выраженная при поражении лицевой части и глазниц. Свод черепа может иметь атрофированные участки, дефекты, узуры, иногда отмечается локальное увеличение костного вещества.
При типе 2 формируются высокодифференцированные опухоли, которые, однако, более агрессивны, чем при заболевании 1 типа. Пигментных пятен нет. Образуются невриномы – подвижные и болезненные неоплазии. Нередко они локализуются на слуховом нерве, вызывая потерю слуха. Нейрофиброматоз 3 типа отличается большим количеством нейрофибром, ускоренным развитием нейролемм и глиом зрительного нерва, приводящих к расстройству зрения. Специфический признак – появление нейрофибром на ладонях. При болезни 4 типа симптомы похожи, сохраняется риск поражения зрительных волокон. Для 5 типа характерны пигментные темные пятна, опухоли больших размеров, провоцирующие асимметрию тела. Течение 6 типа сопровождается лишь пигментными пятнами. При 7 типе выявляются нейрофибромы средних размеров, гиперпигментации нет.
Осложнения
В 10% случаев нейрофибромы трансформируются в злокачественные опухоли. В группе высокого риска находятся пациенты с большим катамнестическим стажем, беременные женщины. У 6% детей нарушается умственное развитие: они имеют проблемы при освоении учебных навыков (чтение, письмо, счет), с трудом запоминают новую информацию, долго адаптируются в незнакомых ситуациях. Больные всех возрастов подвержены депрессии, поскольку испытывают дискомфорт, чувство стыда и неловкости из-за обезображенной внешности. Множественные нейрофибромы провоцируют эндокринные расстройства, эпилептические припадки, гипотонию мышц, стеноз почечной и легочной артерии, легочные кисты, интерстициальную пневмонию, гипертрофию клитора, нарушения развития органов ЖКТ.
Диагностика
Подозрение на нейрофиброматоз возникает при множественных подкожных опухолях, пигментных пятнах, спинальной шванноме, ухудшении слуха и зрения. Обследование проводят дерматовенеролог, невролог, офтальмолог, отоларинголог и генетик. Перед инструментальными и лабораторными процедурами осуществляется сбор семейного и личного анамнеза, клинический опрос и осмотр. В ходе генеалогического анализа выявляется передача заболевания в нескольких поколениях, реже определяется первичная спонтанная мутация. На теле пациентов обнаруживаются нейрофибромы, пигментные области (при определенных типах болезни), искривления позвоночника, деформации костей, нарушения зрения, слуха, координации движений. Производится дифференциальная диагностика различных вариантов нейрофиброматозов, исключается синдром Протея, рассеянный липоматоз, синдром Клиппеля-Треноне-Вебера. Для уточнения диагноза назначаются:
Лечение нейрофиброматозов
В настоящее время терапия данной группы заболеваний заключается в симптоматической помощи больным. Пациенты регулярно проходят обследования, нацеленные на контроль формирования и увеличения опухолей. При наличии нейрофибром, провоцирующих боль, расположенных в местах повышенного риска травмирования, сдавливающих или смещающих жизненно важные органы, проводится их хирургическое удаление. Применяются классические методики резекции неоплазий и участков нервов, криодеструкция, лазерная хирургия. При множественных новообразованиях назначается лучевая терапия, химиотерапия. Больным с поражением опорно-двигательного аппарата показаны реабилитационные мероприятия (физиолечение, ЛФК).
Активно разрабатываются способы этиологического лечения нейрофиброматозов. На стадии клинических испытаний находится терапия ингибиторами RAS (белков-активаторов роста опухолей) у лиц с нейрофиброматозом первого типа. Этап теоретических разработок проходят методы генной инженерии. Усилия ученых-генетиков направлены на создание и внедрение в организм больных нормального НФ1 гена, отвечающего за синтез нейрофибромина, на расшифровку и введение гена ФН2, обеспечивающего транскрипцию белка шванномина. В некоторых медицинских центрах предпринимаются попытки применения патогенетической терапии, в основе которой лежит комплексное использование стабилизаторов мембран тучных клеток, антипролиферативных препаратов и ферментов, корректирующих метаболические процессы.
Прогноз и профилактика
Нейрофиброматозы являются прогностически благоприятными заболеваниями – малигнизация опухолей происходит редко, в большинстве случаев больные остаются трудоспособными и социально адаптированными. При правильных и регулярных реабилитационных мероприятиях нарушения со стороны костной системы и задержка умственного развития успешно корректируются. Поскольку заболевание является наследственным, профилактика возможна на этапе планирования беременности, парам из групп риска (с отягощенным семейным анамнезом) рекомендуется медико-генетическое консультирование с определением вероятности рождения больного ребенка.
Клинико-лучевая диагностика нейрофиброматоза I типа
Распространенность нейрофиброматоза первого типа (НФ1) составляет примерно 1 случай на 3000 человек, однако эта цифра заметно варьирует в зависимости от страны и в России составляет 1 случай на 7 812 человек. Ожидаемая продолжительность жизни людей с нейрофиброматозом первого типа сокращена на
8–21 лет, а причина их смерти чаще всего связана со злокачественными новообразованиями.
НФ1 — генетически обусловленное заболевание с аутосомно-доминантным типом наследования, что означает, что все носители герминативных мутаций в гене 17q11.2 заболеют НФ1. Однако проявления этого заболевания крайне вариабельны и могут различаться даже среди членов одной семьи, имеющих идентичную мутацию. К слову, различных мутаций, обусловливающих заболевание, довольно много (>3000), что связано с большим размером гена (более 60 экзонов) и многообразием типов мутаций (транслокации, делеции, инверсии и точковые мутации), которые могут происходить в этой области — это усложняет генетическую диагностику. Последнюю проводят на материале ДНК лимфоцитов периферической крови и/или опухолевом материале, исключение — сегментарный нейрофиброматоз (иногда называемый нейрофиброматозом V типа), когда поражение затрагивает только один сегмент тела пациента. В последнем случае материалом для диагностики может служить только опухолевый материал.
.
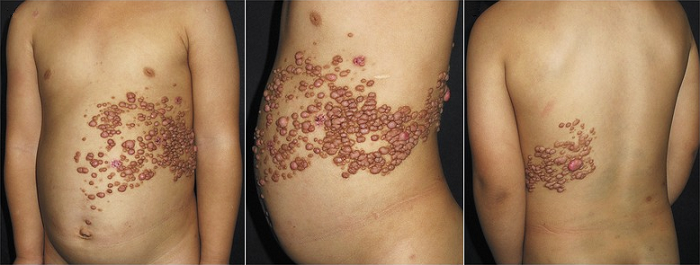
Рисунок 1 | Сегментарный нейрофиброматоз. Множественные нейрофибромы, расположенные в пределах одного или нескольких дерматомов.
Ген 17q11 кодирует белок нейрофибромин, являющийся супрессором опухолевого роста. Нейрофибромин продуцируется в нервных клетках и специализированных клетках нейроглии (олигодендроциты и шванновские клетки) и в норме подавляет протоонкоген RAS, ускоряя его переход в неактивную форму. Невозможность продуцировать функционирующий нейрофибромин приводит к увеличению роста и выживаемости клеток.
Клинические проявления
Пигментные нарушения
Пятна «кофе с молоком» являются одним из наиболее частых кожных симптомов НФ1. Эти пятна могут увеличиваться в размерах и количестве, а также с возрастом приобретать более темную окраску. Начиная с периода новорожденности могут появляться скопления мелких пигментных пятен (веснушек) в подмышечных впадинах, паховой области и в других складках кожи.
Нейрофибромы
Доброкачественные новообразования, характерные для НФ1. Встречаются кожные (подкожные), интраневральные, плексиформные и диффузные нейрофибромы.
.
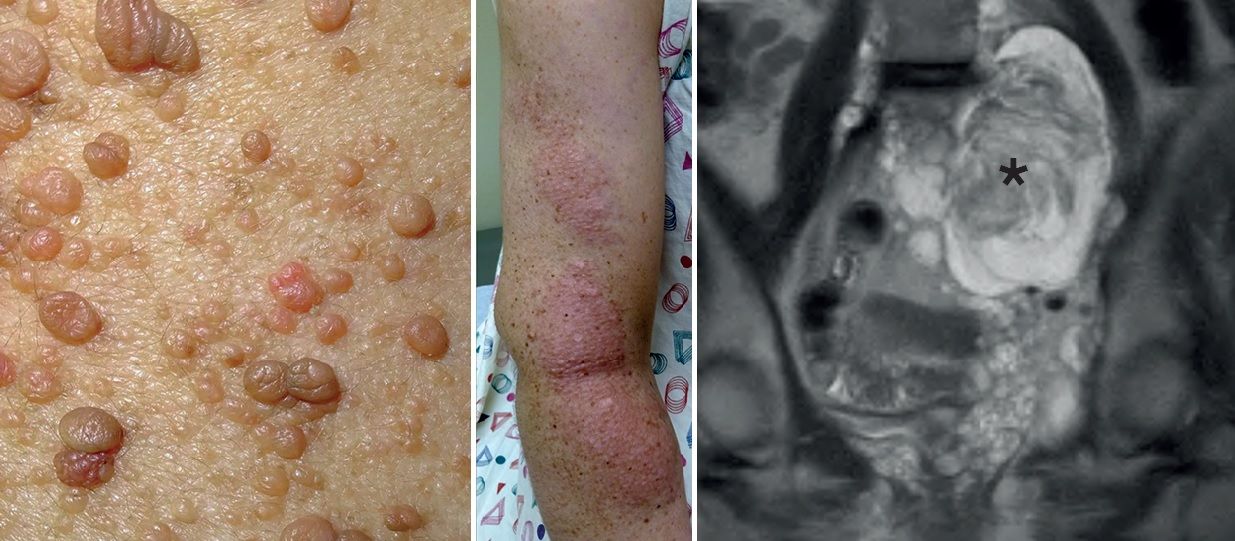
Рисунок 2 | Слева направо: кожные нейрофибромы; интраневральные нейрофибромы по ходу периферического нерва; крупная интраабдоминальная плексиформная нейрофиброма (звездочка).
Кожные нейрофибромы происходят из дермальных клеток-предшественниц, могут быть болезненными или безболезненными узелковыми образованиями, описан симптом «дверного звонка»: при надавливании нейрофиброма полностью проваливается в кожу.
Интраневральные и плексиформные нейрофибромы — это опухолевые образования из оболочек периферических нервов и нервных сплетений. У мышей с двухаллельной потерей гена НФ1 в шванновских клетках удалось смоделировать нейрофибромы, схожие с такими у людей с НФ1. Как и человеческие плексиформные нейрофибромы, опухоли у мышей состояли из различных типов клеток, в том числе тучных клеток, макрофагов, фибробластов, нейронов и шванновских клеток.
Нейрофибромы могут перерождаться в злокачественные опухоли из оболочек нервов, причем наибольший риск представляют плексиформные нейрофибромы, особенно плечевого и пояснично-крестцового сплетений. Также риск перерождения повышен у пациентов с лучевой терапией в анамнезе или с наличием семейной истории малигнизации.
Характерный симптом нейрофибром — образование смещается только латерально, но не вверх-вниз (так как оно «сидит» на нервном стволе). Нейрофибромы могут сдавливать периферический нерв, приводя к потере его функции.
Скелетные аномалии
Для нормального функционирования костной ткани требуется скоординированное взаимодействие между резорбирующими (остеокласты) и костеобразующими (остеобласты) клетками. Использование мышиной модели показало нарушения функции остеобластов (увеличение продукции пирофосфата, снижение костного морфогенетического протеина 2, который способствует дифференциации остеобластов) у мутантных по НФ1 гену мышей. В результате у пациентов отмечается нарушение минерализации костей, дугообразные деформации большеберцовых костей, кифотические и сколиотические деформации, деформации грудной клетки, псевдоартрозы крупных суставов, дисплазия клиновидных костей.
Глиома зрительного тракта
Глиомы — доброкачественные опухоли из зрительного нерва и других частей зрительного анализатора. Расположение может быть любым (орбитальным, интраканальным, интракраниальным — последнее иногда разделяют на пре-, постхиазмальное и хиазмальное), возможно множественное поражение, симметричное расположение опухолей нехарактерно. Показана значительная зависимость роста опухоли от опухолевого окружения, которое представлено в основном клетками нейроглии. Последние влияют на пролиферацию опухолевых клеток путем секреции цитокинов, а также нейротоксинов, которые повреждают аксоны зрительного нерва, приводя к нарушению остроты зрения. Это дает надежду, что в будущем таргетная терапия, нацеленная на опухолевое окружение, сможет замедлять рост глиом зрительного нерва. Данное новообразование редко бывает причиной смерти, но значительно влияет на качество жизни больных. Девочки болеют в 5–10 раз чаще мальчиков.
Кроме того, у больных НФ1 чаще, чем в среднем в популяции, встречаются другие типы опухолей головного мозга (астроцитомы, глиомы).
Другие проявления
Среди опухолевых заболеваний встречаются феохромоцитома, гамартомы радужной оболочки (узелки Лиша).
Часто встречается задержка умственного развития.
Кумулятивный риск злокачественного образования к 50 годам у лиц с нейрофиброматозом типа 1 повышается на 20–39 %, с риском развития рака в течение жизни
Кроме того, люди с нейрофиброматозом типа 1 имеют исключительно высокий риск развития злокачественных опухолей головного мозга (приблизительно в 40 раз выше риск развития глиомы высокой степени злокачественности), эндокринного рака (более чем в 74 раза повышен риск развития рака надпочечников), злокачественных новообразований периферических нервов (> в 1000 раз повышенный риск развития) и рака молочной железы.
Дети с нейрофиброматозом типа 1 имеют повышенный риск лейкемии, острого лимфоцитарного лейкоза, неходжкинской лимфомы.
Кроме того, некоторые исследования подтверждают повышенный риск развития рассеянного склероза, эпилепсии, макроцефалии, гидроцефалии.
.
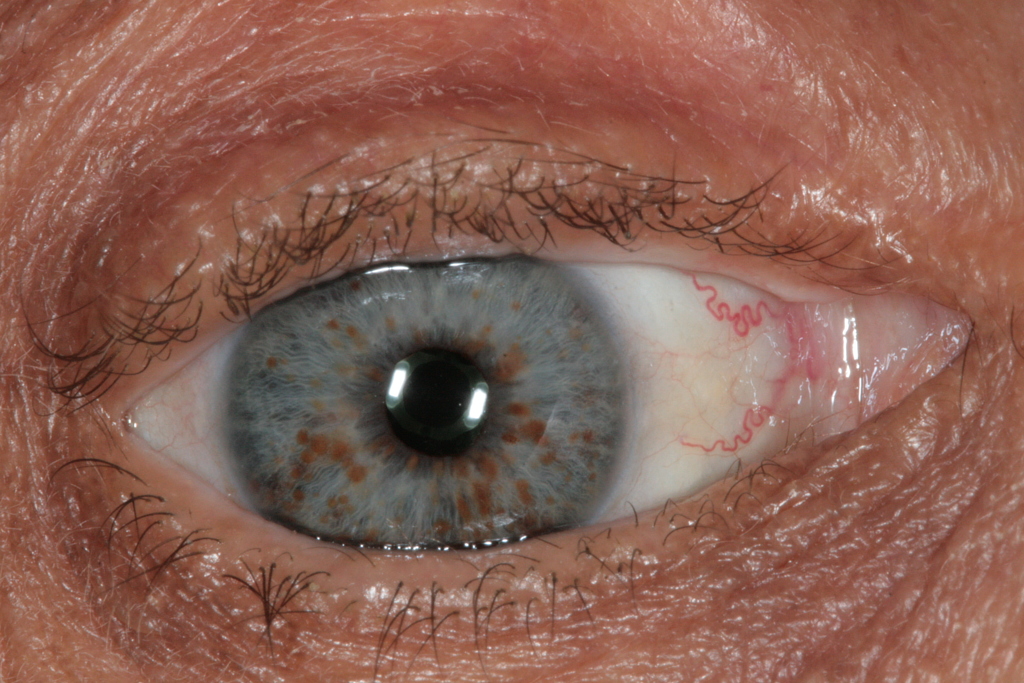
Рисунок 3 | Узелки Лиша.
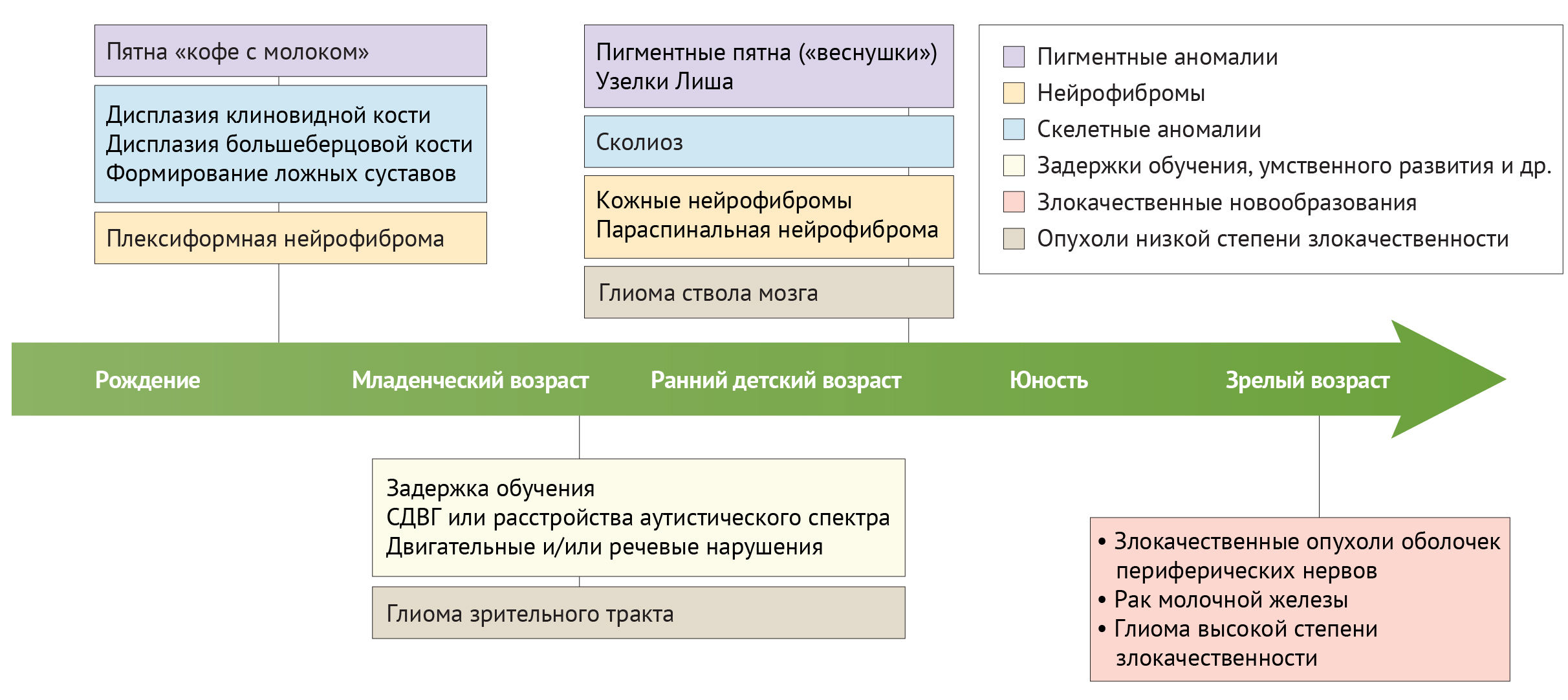
Рисунок 4 | Проявления симптомов нейрофиброматоза первого типа в зависимости от возраста пациента.
Критерием диагностики НФ первого типа считают наличие двух или более признаков (согласно конференции Национального института здоровья по нейрофиброматозу, США, 1988):
Лучевые проявления нейрофиброматоза
Со стороны ЦНС
Глиома зрительного тракта может определяться на КТ (особенно при исследованиях с малой толщиной среза) как веретенообразное или экзофитное расширение зрительного нерва, может также наблюдаться извитость его хода. Однако более чувствительной методикой является МРТ. Глиома проявляет себя высоким в Т2ВИ МР-сигналом (может определяться тонкая гипоинтенсивная полоска по периферии — отодвинутая опухолью твердая мозговая оболочка) и МР-сигналом от изо- до гипоинтенсивного на Т1ВИ; Такие опухоли часто накапливают контрастный препарат.
Дифференциальная диагностика глиомы зрительного нерва в первую очередь проводится с менингиомой: для последней характерно наличие кальцинатов и симптом «трамвайных рельс», когда края сохранного зрительного нерва виднеются в виде параллельных полос на фоне опухоли). Глиому хиазмальной области нужно дифференцировать с ганглионглиомой, менингиомой, астроцитомой или глиомой гипоталамуса и др.
Существует классификация распространенности глиом зрительного тракта по Dodge:
1 стадия: в процесс вовлечен только зрительный нерв.
2 стадия: вовлечение хиазмы.
3 стадия: вовлечение гипоталамуса и/или других прилежащих структур.
.
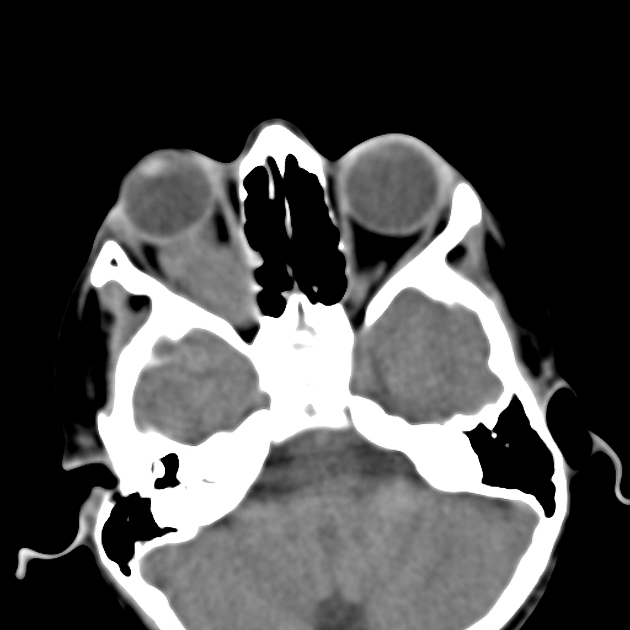
Рисунок 5 | На аксиальном КТ-скане определяется неравномерно расширенный правый зрительный нерв (на скане — слева).
.
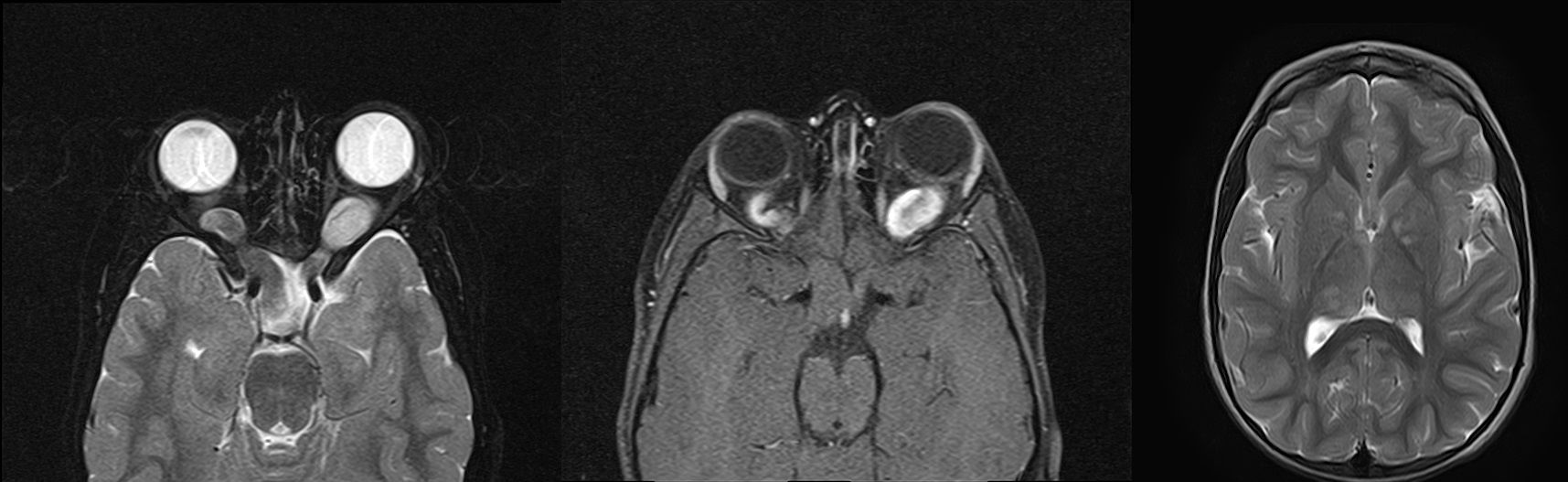
Рисунок 6 | Двусторонние глиомы зрительных нервов у пациента с НФ1. Определяется двустороннее расширение зрительного нерва с повышением МР-сигнала от него на Т2ВИ (1) и интенсивным накоплением контрастного препарата (2). У этого же пациента определялись зоны повышения МР-сигнала в Т2ВИ без четких контуров, масс-эффекта или отека прилежащей паренхимы мозга, расположенные в базальных ядрах и таламусе (3).
Глиомы ствола мозга
Составляют примерно 8–10 % от общего числа новообразований ЦНС при НФ1. Средний возраст пациентов составляет 8 лет. Чаще поражается мозжечок, следующими по частоте являются Варолиев мост и промежуточный мозг. Следует отмечать сопутствующие гидроцефалию и вклинение мозга. Опухоли выглядят гиперинтенсивными на Т2ВИ, обычно гетерогенно накапливают контраст. Чтобы не пропустить слабоинтенсивное контрастное усиление, возможно использование субтракции.
.
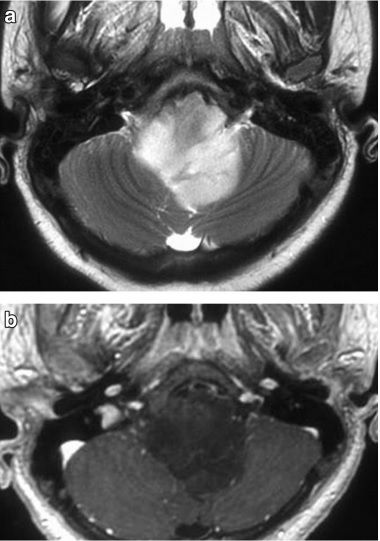
Рисунок 7 | Глиома ствола мозга. В области мозжечка определяется гиперинтенсивное в Т2ВИ (а) образование, которое на постконтастных Т1ВИ (b) демонстрирует слабое негомогенное накопление контраста.
Фокальные области гиперинтенсивности (FASI)
Обнаруживаются у примерно 80 % пациентов с НФ1 и представляют собой области гиперинтенсивного МР-сигнала на Т2ВИ и FLAIR, расположенные в базалных ядрах, таламусе, стволе мозга, мозжечке и субкортикальном белом веществе. Морфологически представляют собой участки миелинопатии с увеличением вакуолей. Корреляция данных изменений с клиникой до конца не понятна.
.
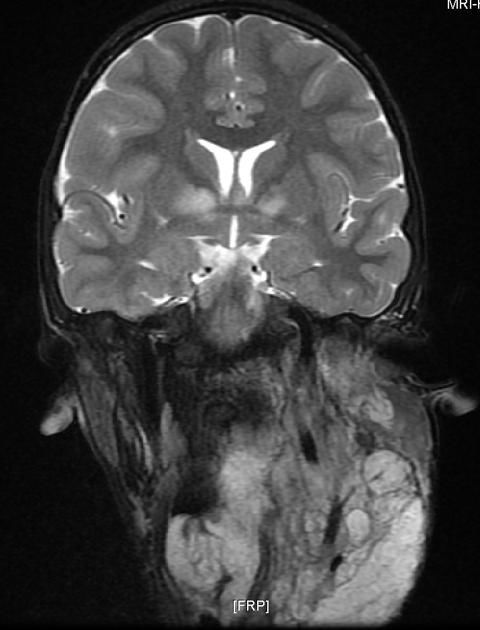
Рисунок 8 | Определяются асимметричные области гиперинтенсивного МР-сигнала в Т2 в базальных ядрах с обеих сторон. Также обращает на себя внимание гиперинтенсивное образование по левой поверхности шеи и головы — плексиформная нейрофиброма.
Другие проявления со стороны ЦНС, области головы и шеи
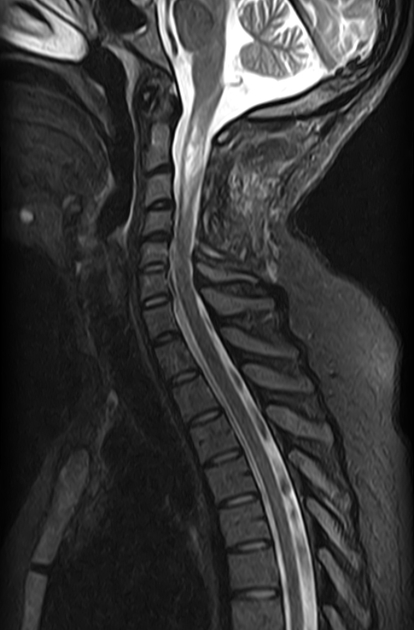
Рисунок 9 | STIR шейного отдела позвоночника в сагиттальной проекции. Определяется гиперинтенсивное интрамедуллярное образование с нечеткими краями — астроцитома спинного мозга. У пациентов с НФ1 повышен риск возникновения злокачественных новообразований ЦНС.
.
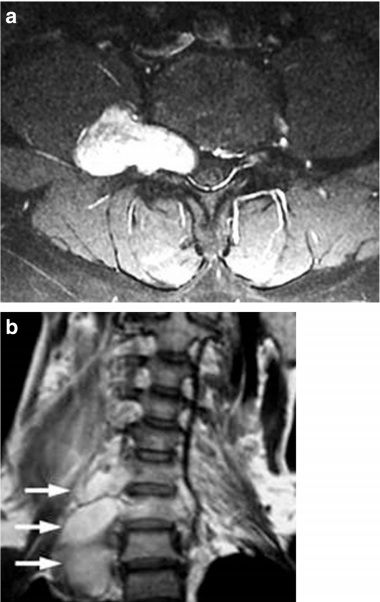
Рисунок 10 | Аксиальный Т2Fs (а) и корональный Т2ВИ (b) у пациента с НФ1. Определяются неравномерное расширение и гиперинтенсивность МР-сигнала от спинномозговых нервов, обусловленные интраневральными нейрофибромами.
.
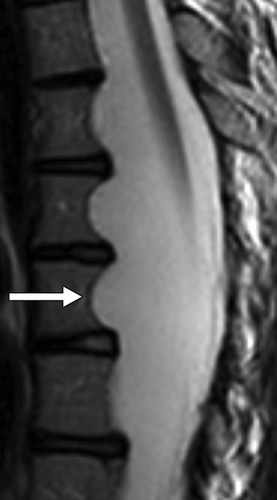
Рисунок 11 | Сагиттальный Т2ВИ поясничного отдела позвоночника. Определяется эктазия (расширение) дурального мешка.
.
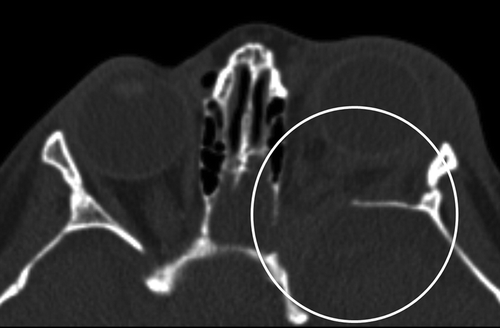
Рисунок 12 | Аксиальный КТ-скан орбит. Определяется дисплазия левого крыла клиновидной кости (кружок); увеличена верхняя орбитальная щель. Нередко в этой области возникают нейрофибромы, приводя к проптозу на стороне поражения.
.
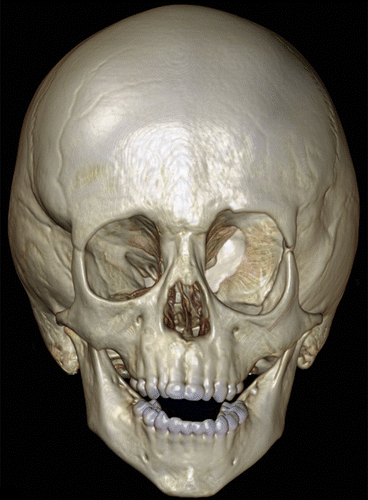
Рисунок 13 | 3D-реконструкция КТ костей черепа. Наблюдается дисплазия левого крыла клиновидной кости.Также обращает на себя внимание увеличение мозгового черепа.
Костно-мышечные проявления НФ1
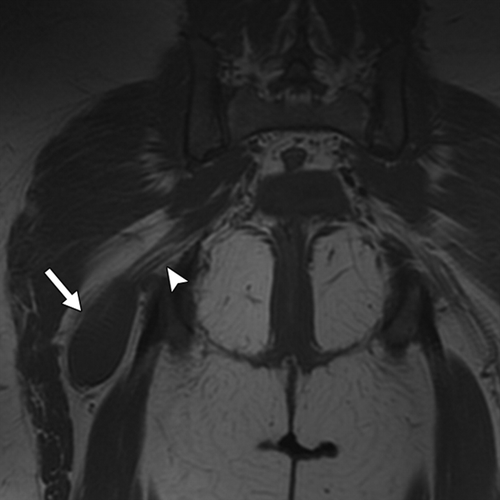
Рисунок 14 | Аксиальный Т1ВИ области таза. В области правого седалищного нерва определяется округлое образование изоинтенсивного с мышцами МР-сигнала — солитарная интраневральная нейрофиброма.
.
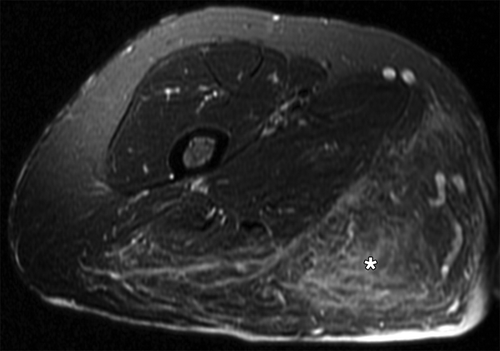
Рисунок 15 | Аксиальный PDFs скан правого бедра. Определяется область гиперинтенсивного сигнала в подкожной жировой клетчатке (звездочка) — диффузная нейрофиброма.
.
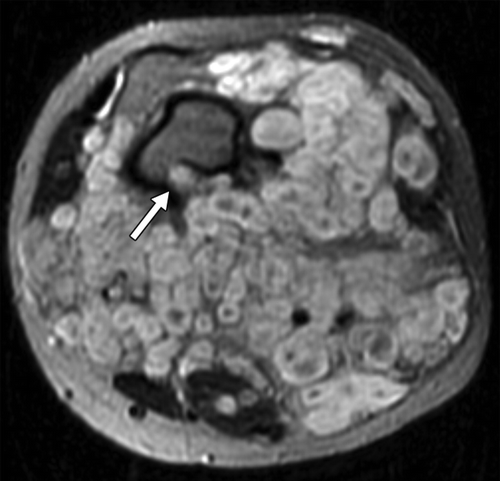
Рисунок 16 | Аксиальное Т2ВИ левого бедра. Множественные узелки гиперинтенсивного МР-сигнала с гипоинтенсивной областью в центре (сиптом мишени). Обратите внимание на масс-эффект и эрозирование кортикального слоя кости (стрелка). Плексиформная нейрофиброма.
.
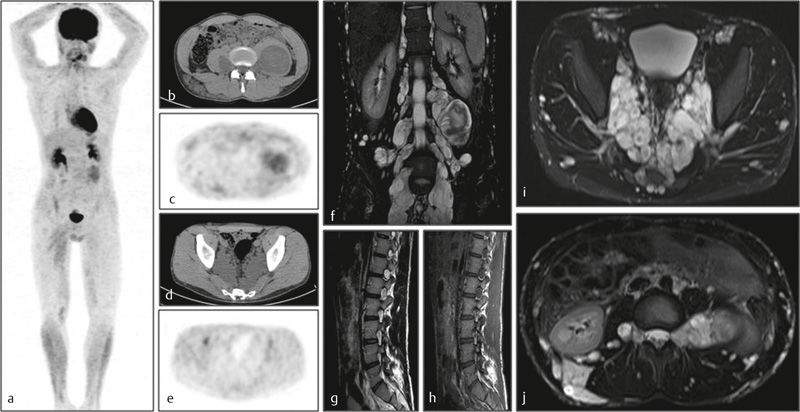
Рисунок 17 | Пациентка 20 лет с множественными нейрофибромами. 18F-FDG ПЭТ (а) не показала очагов патологического метаболизма глюкозы; b,c — нейрофиброма левой подвздошно-поясничной мышцы с умеренным повышением метаболизма; d,e — гиперденсная нейрофиброма левой подвздошной области без повышения метаболизма; f-j — множественные спинальные нейрофибромы с гиперинтенсивным МР сигналом в Т2ВИ.
.
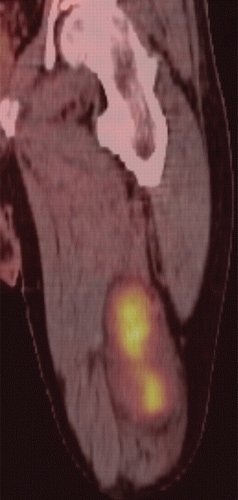
Рисунок 18 | 18F-FDG ПЭТ/КТ изображение показало объемное новообразование с интенсивным повышением метаболизма — злокачественная опухоль из оболочки периферического нерва. 18F-FDG ПЭТ является наиболее чувствительным методом дифференциальной диагностики злокачественных опухолей из оболочек периферических нервов с нейрофибромами.
.
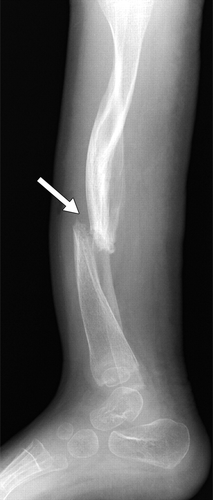
Рисунок 19 | Ложный сустав и дугообразная деформация обеих берцовых костей.
.
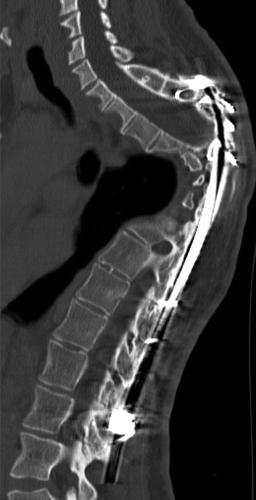
Рисунок 20 | КТ в сагиттальной реконструкции. Выраженная кифотическая деформация, дистрофический сколиоз. Определяются артефакты от металлической фиксирующей конструкции.
Источники: