НЕЙРОФИБРОМАТОЗ I ТИПА. Проблемы диагностики и лечения
Каковы диагностические критерии нейрофиброматоза? Что является особенностью нейрофиброматоза и затрудняет диагностику? На чем основывается терапия нейрофиброматоза? Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингха
Каковы диагностические критерии нейрофиброматоза?
Что является особенностью нейрофиброматоза и затрудняет диагностику?
На чем основывается терапия нейрофиброматоза?
 Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингхаузена) — это тяжелое системное наследственное заболевание с преимущественным поражением кожи и нервной системы, одно из наиболее распространенных моногенных заболеваний человека, встречающееся с частотой не реже 1:3000 — 1:4000 населения. Наследуется аутосомно-доминантно, с высокой пенетрантностью и вариабельной экспрессивностью. Заболевание обусловлено мутацией гена «нф1» в 17q-хромосоме. Мужчины и женщины поражаются одинаково часто. Примерно половина случаев — следствие новых мутаций.
Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингхаузена) — это тяжелое системное наследственное заболевание с преимущественным поражением кожи и нервной системы, одно из наиболее распространенных моногенных заболеваний человека, встречающееся с частотой не реже 1:3000 — 1:4000 населения. Наследуется аутосомно-доминантно, с высокой пенетрантностью и вариабельной экспрессивностью. Заболевание обусловлено мутацией гена «нф1» в 17q-хромосоме. Мужчины и женщины поражаются одинаково часто. Примерно половина случаев — следствие новых мутаций.
Заболевание характеризуется выраженным клиническим полиморфизмом, прогрессирующим течением, полиорганностью поражений и высокой частотой осложнений, в том числе приводящих к летальному исходу (развитие сердечно-легочной недостаточности вследствие выраженных скелетных аномалий, злокачественное перерождение нейрофибром и др.).
Механизм развития клинических проявлений неизвестен. Существует предположение, что ген «нф 1» входит в группу генов, подавляющих рост опухолей. Снижение или отсутствие выработки продукта гена — нейрофибромина приводит к диспластической или неопластической пролиферации клеток.
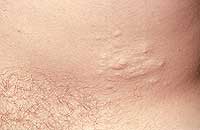
Клиническая диагностика нейрофиброматоза I типа основывается на обнаружении диагностических критериев, рекомендованных Международным комитетом экспертов по нейрофиброматозу. Диагноз может быть поставлен при наличии у больного по крайней мере двух из перечисленных ниже признаков: не менее пяти пятен цвета «кофе с молоком» диаметром более 5 мм у детей препубертатного возраста и не менее шести таких пятен диаметром более 15 мм в постпубертатном периоде; две и более нейрофибромы любого типа или одна плексиформная нейрофиброма; множественные мелкие пигментные пятна типа веснушек, локализованные в крупных кожных складках (подмышечных и/или паховых); глиома зрительного нерва; два и более узелков Лиша на радужной оболочке, обнаруживаемых при исследовании с помощью щелевой лампы; дисплазия крыла клиновидной кости или врожденное истончение кортикального слоя длинных трубчатых костей с наличием псевдоартроза или без него; наличие у родственников первой степени родства нейрофиброматоза I типа по тем же критериям.
 |
| Рисунок 1. Множественные нейрофибромы |
Особенностью заболевания является специфическая последовательность проявления симптомов в зависимости от возраста пациента, что затрудняет клиническую диагностику нейрофиброматоза I типа в раннем детском возрасте. Таким образом, с рождения или первых лет жизни могут существовать лишь некоторые признаки нейрофиброматоза I типа, такие как крупные пигментные пятна, плексиформные нейрофибромы, скелетные дисплазии. Другие симптомы могут проявиться значительно позднее (к 5–15 годам). При этом степень выраженности клинических проявлений, течение и быстрота прогрессирования нейрофиброматоза I типа у разных больных неодинаковы и колеблются в широких пределах. В настоящее время не установлено, чем обусловлены такие различия.
Нейрофибромы (дермальные, гиподермальные, плексиформные) представляют собой наиболее выраженное проявление болезни Реклингхаузена, их количество иногда достигает нескольких тысяч; плексиформные нейрофибромы могут быть гигантскими, массой более 10 кг. Эти косметические дефекты, как правило, больше всего беспокоят пациентов, даже имеющих системные заболевания. Кроме того, нейрофибромы, особенно плексиформные, связаны с повышенным риском озлокачествления (в 20% случаев, по нашим данным). При локализации в средостении, в брюшной полости, в глазнице они приводят к нарушению функций прилегающих органов. Например, в сентябре 2000 года в отделении наследственных заболеваний кожи ЦНИКВИ был консультирован больной мальчик 8 лет, прибывший из Брянской области в Москву для оперативного лечения по жизненным показаниям; гигантская плексиформная нейрофиброма располагалась в верхнем средостении, деформировала нижнюю 1/3 шеи и являлась причиной затрудненного дыхания и пароксизмальной тахиаритмии.
О развитии нейрофибром известно немногое. Время от времени растет их количество и размеры в ответ на различные стимулы, среди которых ведущее место занимают гормональная перестройка организма: пубертатный возраст, период беременности или после родов, а также перенесенные травмы или тяжелые соматические заболевания. С расширением спектра предлагаемых коммерческих медицинских и косметических услуг значительно увеличилось число обращений больных, указывающих на появление новых опухолей (нейрофибром, неврином, шванном) после ятрогенных вмешательств. Речь идет об удалении опухолей с диагностической или лечебной целью различными методами, в том числе с помощью хирургического иссечения. К ятрогенным осложнениям также приводит назначение физиотерапевтических процедур при лечении различных соматических заболеваний, коррекции скелетных нарушений (всевозможных видов сколиоза, переломов) и нервно-мышечных расстройств (очень часто массаж по различным поводам назначается детям грудного возраста, когда диагностика нейрофиброматоза I типа зачастую невозможна из-за недостаточности клинических проявлений). Но часто заболевание прогрессирует и на фоне кажущегося благополучия. Положение осложняется тем, что у врачей нет возможностей приостановить развитие болезни.
Основной задачей научных исследований представляется разработка методов патогенетического лечения нейрофиброматоза I типа, позволяющих сдерживать появление новых и рост уже имеющихся опухолей, а также предотвращать развитие осложнений.
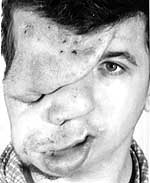 |
| Рисунок 2. Плексиформная нейрофиброма |
В настоящее время для лечения этого заболевания как за рубежом, так и в России используются методы симптоматической терапии, как, например, хирургическое удаление опухолей, коррекция кифосколиоза или лучевая терапия нейрофибром внутренних органов. Кроме того, ученые на Западе сконцентрировались на возможности этиологического лечения, то есть генной инженерии. Особенно далеко это направление продвинулось со времени открытия мутантного гена и расшифровки его первичного продукта — нейрофибромина в 1990 году; на научные изыскания, связанные с этой проблемой, ежегодно выделяются огромные средства.
Первая попытка патогенетического подхода к лечению была сделана V. Riccardi в 1987 г., когда он предложил длительное использование кетотифена (в дозе 2-4 мг в течение 1,5-3 лет) для стабилизации мембран тучных клеток, полагая, что именно дегрануляция этих клеток стимулирует рост опухолей. Однако лечение одним кетотифеном не принесло желаемых результатов: уменьшались субъективные ощущения болезненности и зуда в области нейрофибром, но какого-либо влияния на рост опухолей отмечено не было. Кроме того, при продолжительном приеме препарата наблюдалось снижение количества иммунокомпетентных клеток в периферической крови и ухудшение показателей иммунитета. Существуют различные точки зрения на роль тканевых базофилов в развитии нейрофибром. По мнению некоторых авторов, тучные клетки представляют собой эффекторные клетки противоопухолевого иммунитета. С помощью обычной и электронной микроскопии было показано, что большое количество тканевых базофилов с их активной внеклеточной дегрануляцией наблюдается только на ранней стадии развития нейрофибром. На поздней же стадии, при длительности существования нейрофибром не менее пяти лет, в клеточном матриксе опухолей тучных клеток значительно меньше, дегрануляция их преимущественно внутриклеточная и не сопровождается разрушением клеток.
На основании данных многочисленных исследований нами впервые разработана комплексная методика патогенетической терапии с использованием препаратов из разных групп лекарственных средств. Учитывая то, что клеточный состав нейрофибром в основном представлен шванновскими клетками, фибробластами, тучными клетками и лимфоцитами, а межклеточное вещество в активно растущих, особенно плексиформных, — кислыми мукополисахаридами, для лечения нейрофиброматоза I типа мы выбрали следующие препараты. Стабилизатор мембран тучных клеток, кетотифен, мы назначали по 2-4 мг короткими курсами по два месяца. Чтобы избежать осложнений, в первые две недели приема препарата использовался фенкарол по 10-25 мг три раза в день. В качестве антипролиферативного препарата применялись тигазон в дозе не менее 1 мг на килограмм массы тела или аевит до 600 000 МЕ с учетом переносимости. Также курсами применялась лидаза (мукополисахаридаза) в дозе 32-64 Ед в зависимости от возраста внутримышечно, через день, на курс 30 инъекций.
Вышеуказанные препараты использовались комплексно в различных сочетаниях или в виде монотерапии в зависимости от формы нейрофиброматоза I типа, жалоб, течения, а также возраста и пола больных. Обязательно лечение проводилось в периоды прогрессирования заболевания, то есть при появлении новых опухолей и/или росте уже имеющихся, как правило сопровождающемся зудом или ощущением болезненности в их проекции, а также с целью предотвращения активизации заболевания во время планируемых операций на опухолях. Повторные курсы лечения с интервалами в два месяца назначались при наличии у больных крупных плексиформных нейрофибром или болезненных неврином. При этом, как правило, курсовое применение тигазона (или аевита) в виде монотерапии чередовалось с сочетанным применением стабилизаторов мембран тучных клеток и инъекций лидазы. Предлагаемое лечение хорошо переносилось больными.
В единичных случаях наблюдались незначительное повышение уровней печеночных показателей в повторных биохимических анализах крови при приеме тигазона (у одной больной) и очаговая аллергическая реакция на введение лидазы (у двух пациентов из 60), которая проявлялась воспалением тканей в месте инъекций. В этих случаях препараты отменялись, назначалось симптоматическое лечение.
В результате проводимой терапии нам, как правило, удавалось приостановить прогрессирование заболевания; наблюдалось уменьшение (сморщивание) нейрофибром и неврином вплоть до полного исчезновения некоторых опухолей (особенно активно уменьшаются плексиформные нейрофибромы — на ранней стадии своего развития — и невриномы).
Полученные результаты удовлетворяют исследователей и позволяют рекомендовать вышеуказанную методику патогенетического лечения нейрофиброматоза I типа для повсеместного применения. Разработанная нами комплексная методика патогенетического лечения впервые дает возможность оказать больным медикаментозную помощь.
Нейрофиброматоз диагностические критерии у взрослых что это
а) Синоним. Нейрофиброматоз 1 типа (НФ1) также известен как периферический нейрофиброматоз (НФ) или болезнь фон Реклингхаузена.
б) Эпидемиология. Наиболее распространенная форма нейрофиброматоза (НФ) составляет около 96% случаев с заболеваемостью I на 4000 живорожденных. Гендерное и расовое преобладание не описано.
в) Этиология. Нейрофиброматоз 1 типа (НФ1) наследуется по аутосомно-доминантному типу почти с полной пенетрантностью, но варьирующей экспрессией. Тем не менее, положительный семейный анамнез отмечается почти у 50% пациентов, остальные 50% случаев, как полагают, представляют собой новые мутации. Участвует ген супрессоров опухолей, Ген расположен в перицентрической области на длинном плече 17-й хромосомы и имеет чрезвычайно высокую скорость спонтанной мутации более чем с 200 вариантами, известными на настоящий момент.
Продукт гена, нейрофибромин, представляет собой большой цитоплазматический белок, существующий в нескольких формах в различных тканях; он, как полагают, взаимодействует с внутриклеточными цитоплазматическими микротрубочками.
г) Диагностика и лечение нейрофиброматоза 1 типа. Из-за отсутствия общепринятого генетического теста диагноз основан на клинических проявлениях. Они не всегда присутствуют с рождения, проявляясь в разные периоды жизни.
2. Глазные проявления:
— Узелки Пиша: пигментированные (желто-коричневые), гамартомы радужки, легко обнаружить с помощью щелевой лампы. Часто отсутствуют в детстве, узелки Лиша появляются с возрастом и имеются примерно у 94% постпубертатных пациентов. Гистопатологически они представлены массой меланоцитов.
— Другие (необычные) глазные проявления: врожденная глаукома, нейрофибромы век и конъюнктивы, утолщение роговицы, конъюнктивы и цилиарного нерва, астроцитомы сетчатки.
3. Опорно-двигательные нарушения:
— Клиновидная дисплазия крыла основной кости: наиболее типичное костное поражение, приводит к пульсирующему экзофтальму.
— Сколиоз: встречается у 10-20% пациентов, часто проявляется в подростковом возрасте.
— Большеберцовой ложный сустав: может быть устойчивым к лечению с необходимостью ампутации в 80% случаев.
— Другие: истончение коркового слоя кости, гиперплазии конечностей, и (редко) рабдомиосаркома.
4. Неврологические проявления:
— Периферические нейрофибромы: множественные кожные, подкожные и периферические поражения, часто распространяются в торакоабдоминальной области или вокруг соска. Гистологически доброкачественные (злокачественные преобразования редки и могут быть представлены в количестве от нескольких до нескольких тысяч). В основном они представляют собой косметическую проблему с возможностью определенной хирургической коррекции по желанию пациента.
— Плексиформные нейрофибромы: часто поражают большое количество периферических нервов или часть симпатической цепочки с потенциальным обезображиванием (гемигипертрофия руки/ноги) или нарушением функции вовлеченных областей. В целом доброкачественные, но злокачественное преобразование происходит (злокачественная оболочки периферических нервов опухоль: MPNST) в 6% случаев. Для бессимптомных поражений предпочтительна тактика выжидания и наблюдения; результаты лечения с 13-цис-ретиноевой кислотой и интерфероном альфа-2а находятся в стадии оценки.
При симптоматическом течении возможно хирургическое уменьшение/удаление опухоли. Тем не менее, риск местного рецидива высок; помимо этого, многие симптоматические поражения порой включают несколько невральных пучков с высоким риском послеоперационного неврологического дефицита. Для MPNST рекомендуется биопсия с последующей возможной ампутацией конечностей, лучевой терапией и химиотерапией (5-летняя выживаемость: 40%).
— Параспинальные нейрофибромы: наиболее распространенные опухоли, поражающие позвоночник у пациентов с нейрофиброматозом 1 типа (НФ1). Параспинальные нейрофибромы, как правило, возникают из спинальных корешков в шейном и поясничном отделах, они могут врасти в позвоночный канал через межпозвонковые отверстия, в результате чего формируются гантелевидные поражения. Хирургическая резекция показана во всех симптоматических случаях и по рентгенологическим критериям (масс-эффект) для шейно-грудной области (риск вторичной миелопатии в связи со сдавлением спинного мозга).
— Глиомы зрительных путей: наиболее распространенное проявление в центральной нервной системе при нейрофиброматозе 1 типа (НФ1), поражает примерно 15% пациентов. Эта опухоль обычно встречается в детском возрасте с наибольшим риском возникновения в течение первых шести лет жизни; женщины страдают вдвое чаще, чем мужчины. Развитие возможно в любой точке зрительного пути, но чаще всего встречаются прехиазмальные поражения. Приблизительно половина всех оптических глиом протекает бессимптомно, и большинство из них, в том числе симптоматические, редко прогрессируют.
Глазные признаки являются наиболее частыми клиническими проявлениями; у 39% детей с поражениями хиазмы выявляется гипоталамо-гипофизарная дисфункция.
д) Рекомендации:
— Бессимптомные случаи: регулярный офтальмологический осмотр, ежегодно в возрасте до шести лет. При выявлении аномалий проводится МРТ мозга и глазниц.
— Симптоматические случаи: обязательно постоянное наблюдение с офтальмологическими осмотрами и МРТ с контрастным усилением каждые три месяца в течение первых 18 месяцев, с последующими контрольными наблюдениями через определенные интервалы при стабильном состоянии.
Хирургическое лечение ограничено определенными показаниями: у детей с внутриглазничным и прехиазмальным поражениями и закрывающимися глазами (или проптоз глаза с тяжелыми формами нарушения зрения) операция может быть выполнена в косметических целях и для предотвращения распространение опухоли на хиазму. При хиазмальных опухолях вмешательство может потребоваться для уменьшения объема больших опухолей, особенно с кистозным компонентом.
У детей старше трех лет лучевая терапия является вариантом лечения, почти 80% случаев дают стабилизацию/уменьшение опухоли. Тем не менее, и в таких случаях при лучевой терапии существует риск нейрокогнигивных и нейроэндокринных последствий, помимо вызываемых облучением злокачественных новообразований. За последние 15 лет ряд клинических исследований показал, что прогрессирование опухоли может быть приостановлено при использовании одно- и мультиагентной химиотерапии, и, следовательно, химиотерапия в настоящее время предлагается в качест ве терапии первой линии, особенно у более молодых пациентов (моложе трех лет), для которых лучевая терапия противопоказана.
— Аномалии ликворных пространств: некоторое расширение желудочков относительно часто выявляется при нейрофиброматозе 1 типа (НФ1), но его роль в патогенезе задержки психомоторного развития спорна. Действительно, в большинстве случаев расширение желудочковой системы не связано с повышением внутричерепного давления и не является прогрессирующим. Это относится и к расширению субарахноидальных влагалищ корешков спинного мозга. Лишь в исключительных случаях эти менингеальные растяжения могут стать симптоматическими при сжатии или деформации корешков спинного мозга, следовательно, будет необходимо лечение (как правило, шунтирование).
— Другие внутричерепные опухоли: глиомы полушарий встречаются менее чем в 0,5%, а глиомы задней ямки менее, чем в 1% случаев нейрофиброматоза 1 типа (НФ1). Большинство этих опухолей не имеет тенденции к росту. По этой причине, как и для поражений зрительных путей, последовательный мониторинг (клинический и рентгенологический) целесообразен в бессимптомных случаях, а оперативное вмешательство показано для пациентов с симптомами и/или опухолями с тенденцией к росту. Неопознанные яркие включения при МРТ — частое явление у больных с НФ1.
Они представлены областями спонтанной гиперинтенсивности на Т2-взвешенных изображениях, чаще всего с участием базальных ганглиев, в мозжечке, в стволе головного мозга и подкорковом белом веществе. Предполагается, что они представляют собой участки миелинопатии, которые, как правило, разрешаются спонтанно.
— Неврологическая задержка развития: 30-65% пациентов с нейрофиброматозом 1 типа (НФ1) проявляют некоторую степень неспособности к обучению, речевые навыки сохраняются лучше, чем визуальнопространственные.
е) Прогноз. При самом лучшем медицинском обслуживании продолжительности жизни при нейрофиброматозе 1 типа (НФ1) остается примерно на 15 лет меньше, чем у населения в целом. Озлокачествление является одной из основных причин снижения ожидаемой продолжительности жизни у взрослых с НФ1, а общий риск злокачественных осложнений составляет примерно 3-15%.
 Национальный институт здоровья (NIH),
Национальный институт здоровья (NIH),
итоговая конференция по критериям диагностики для нейрофиброматоза (НФ1);
время появления клинических признаков. 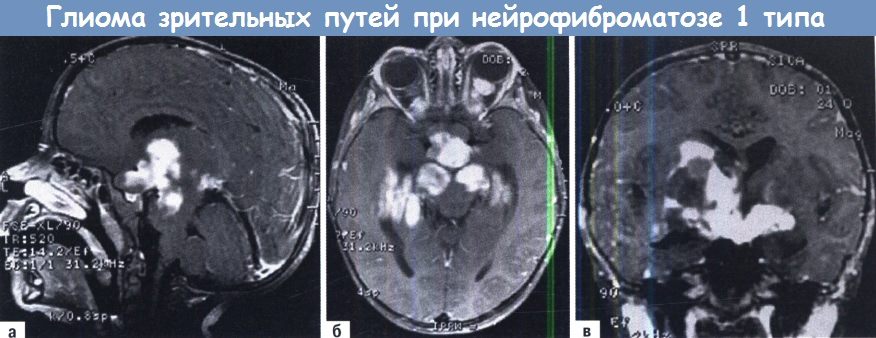 A-В Сагиттальная (А), аксиальная (Б) и коронарная (В) МРТ в режиме Т1 после введения гадолиния 6-месячному ребенку с нейрофиброматозом 1 типа (НФ1) и диффузной глиомой зрительных путей.
A-В Сагиттальная (А), аксиальная (Б) и коронарная (В) МРТ в режиме Т1 после введения гадолиния 6-месячному ребенку с нейрофиброматозом 1 типа (НФ1) и диффузной глиомой зрительных путей.
Как это часто случается у такого рода пациентов, рост опухоли вовлек оба зрительных нерва, хиазму и зрительные тракты.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Содержание
Дерматология в России
Зарегистрируйтесь!
Если Вы врач, то после регистрации на сайте Вы получите доступ к специальной информации.
Если Вы уже зарегистрированы, введите имя и пароль (форма в верхнем правом углу или здесь).
Нейрофиброматоз: обзор, посвящённый NF1, NF2 и шванноматозу
Нейрофиброматоз: обзор, посвящённый NF1, NF2 и шванноматозу
Нейрофиброматоз – гетерогенная группа наследственных онкологических синдромов, приводящих к опухолям центральной и периферической нервной систем.
На сегодняшний день наиболее распространенной формой является нейрофиброматоз 1-го типа (NF1, 96%), за которым следует нейрофиброматоз 2-го типа (NF2, 3%) и менее известная форма – шванноматоз. Нейрофиброматоз не имеет половой или расовой предрасположенности.
Нейрофиброматоз 1 типа
Введение. NF1, также известный как болезнь фон Реклингхаузена или периферический нейрофиброматоз, является синдромом аутосомно-доминантной предрасположенности к опухоли, наиболее часто характеризующимся развитием множественных нейрофибром периферических нервов.
Ожидаемая продолжительность жизни сокращается в среднем до 54 лет, часто из-за злокачественных новообразований (2).
Злокачественные новообразования, связанные с NF1, включают злокачественные опухоли оболочек периферических нервов, глиомы, лейкемию, феохромоцитомы, стромальные опухоли желудочно-кишечного тракта ( GI ) и другие. NF1 вызван мутацией в гене-супрессоре опухоли нейрофибромина, расположенном на хромосоме 17.
Он имеет высокую пенетрантность, а частота мутаций гена NF высока, причем 80% имеют наследование по линии отца (3). Самое раннее известное изображение предполагаемого нейрофиброматоза датируется 13 веком с рисунками цистерцианского монаха (4).
В 1862 году Вирхов сослался на наследственный компонент, когда описал человека, у которого “тело было полностью покрыто шишками размером от булавочной головки до голубиного яйца”, и он отметил, что “проявления наследовались уже в течение трех поколений».
Однако именно студент Вирхова Фридрих фон Реклингхаузен в 1882 году дал наиболее полное клиническое и гистологическое описание этого заболевания и ввел термин “нейрофиброма”.
Диагностические критерии. Симптомы NF1 могут значительно различаться у пациентов, и поэтому своевременный и конкретный диагноз трудно установить.
Хотя клинический диагноз может быть заподозрен очень рано в детстве или младенчестве, основные признаки могут полностью отсутствовать до более старшего возраста.
Приблизительно 30% пациентов с NF1 будут соответствовать одному из приведенных ниже критериев к возрасту 1 год, 97% пациентов будут соответствовать двум критериям к возрасту 8 лет; в ретроспективном обзоре пациентов с NF1, все пациенты соответствовали критериям к возрасту 20 лет (1).
В 1987 году Национальные институты здравоохранения (NIH) США разработали консенсус в отношении диагноза из-за клинических различий.
Клинический диагноз может быть установлен, если критерии, перечисленные в таблице 1, удовлетворяются без возможного альтернативного диагноза.
Патогенез и генетические особенности. Ген, ассоциированный с NF1, был идентифицирован в 1990 году, и было обнаружено, что он является одним из крупнейших генов в геноме человека, охватывающим 280 kbp ДНК (6). Ген NF1 расположен на хромосоме 17q11.2, которая кодирует белок, известный как нейрофибромин (7).
Нейрофибромин относится к активирующему GTPase семейству белков-супрессоров опухоли, которые регулируют функцию передачи сигналов RAS/MAPK и механистическую мишень рапамицина (mTOR). Примерно 50% мутаций происходят de novo у пациентов без семейного анамнеза.
Для семей с унаследованной мутацией наблюдается полная пенетрантность; однако клинические проявления у членов семьи могут сильно различаться. Вариабельность фенотипической экспрессии, вероятно, является результатом эпигенетической модификации (8).
Исследования, посвященные корреляции генотипа и фенотипа, показали, что делеция всего гена, известная как микроделеция 17q11.2, связана с более тяжелой формой заболевания, в то время как мозаицизм может привести к легкому или даже сегментарному изменению (6, 7).
Было идентифицировано почти 1500 различных мутаций гена NF1, и эти мутации были обнаружены по всей длине большого локуса гена (6).
Нонсенс, сдвиг рамки считывания и точечные мутации были идентифицированы с большинством мутаций, приводящих к укороченной форме белка.
Генетическое тестирование часто проводится у членов семьи пациента, у которого была выявлена патологическая мутация, и, таким образом, конкретная мутация может быть проверена у их родственников.
В противном случае, учитывая удивительно высокую степень подверженности мутациям и отсутствие “горячих точек” на гене, NF1 не поддается мутационному анализу в качестве практического диагностического инструмента (6).
Диагноз NF1 чаще всего ставится на основании клинических данных, рассмотренных выше.
Клинические проявления
Нейрофибромы. Нейрофибромы являются наиболее распространенным типом опухолей NF1, встречающейся примерно у 60% пациентов. Гистологически нейрофибромы NF1 неотличимы от спорадических опухолей, хотя первые часто бывают крупнее.
Нейрофибромы могут быть кожными или внутренними, затрагивающими глубокие мягкие ткани. Кожные формы могут быть пятнистыми, узловатыми или бляшкообразными, развивающимися в позднем детстве и увеличивающимися в количестве во взрослом возрасте (2).
Внутренние или глубокие нейрофибромы могут возникать по всему телу, включая периорбитальные, забрюшинные области, желудочно-кишечный тракт и средостение (9).
Патогномоничные для NF-1 плексиформные нейрофибромы – это внутренние нейрофибромы, которые вместо того, чтобы расти интраневрально в пределах одного нерва, разрастаются, вовлекая несколько пучков или ветвей нерва или сплетения.
Эта модель роста соответствует характерному описанию “мешка с червями”, данному при пальпации или хирургическом исследовании этих опухолей. Плексиформные нейрофибромы часто развиваются в детстве и быстро растут, оказывая массовое воздействие на соседние структуры.
В отличие от кожной формы, плексиформные нейрофибромы имеют повышенный риск трансформации в злокачественные опухоли оболочек периферических нервов (MPNST). MPNST являются редкими агрессивными веретеноклеточными саркомами, на долю которых приходится лишь около 5% всех сарком мягких тканей.
Около 50% MPNST развивается при NF1, поскольку у этих пациентов риск развития одного из них в течение жизни составляет от 8 до 13% (3).
Внезапное изменение или рост плексиформной нейрофибромы при наблюдении за визуализацией, а также повышенное поглощение фтордезоксиглюкозо-позитронно-эмиссионной томографии (ПЭТ-сканирование) должны вызывать подозрение на злокачественную трансформацию.
Лечение нейрофибром включает хирургическую резекцию и лазерную терапию, в то время как цель хирургии плексиформной нейрофибромы часто заключается в ее удалении. Недавние клинические испытания с участием ингибитора тирозинкиназы иматиниба показали уменьшение объема опухоли более чем на 20% у подгруппы пациентов (10).
В настоящее время проводятся дальнейшие исследования, посвященные этой терапии, а также ингибиторам mTOR (10).
MPNST являются значительной причиной смертности у пациентов с NF1, и, несмотря на радикальное иссечение с широким хирургическим доступом с последующим химиолучевым лечением, 5-летняя выживаемость остается низкой из-за частых метастазов в легких и костях, а также местных рецидивов (2).
Пигментные аномалии. Пятна кофе с молоком – это доброкачественные коричнево-бежевые пигментированные макулы, которые могут встречаться в любом месте тела и часто являются признаком NF1. К возрасту 1 года у 99% пациентов с диагнозом NF1 будет шесть или более макул цвета кофе с молоком размером более 5 мм (препубертатные критерии в соответствии с NIH) (1, 9).
Хотя пятна с кофе с молоком являются общей особенностью NF1, они неспецифичны, поскольку их можно увидеть примерно у 10% населения в целом, а также при других генетических синдромах – карликовости Сильвера Рассела, MEN IIb, синдроме Легиуса и синдроме Маккьюна-Олбрайта.
Количественное ограничение, используемое в диагностических критериях, основано на исследовании Кроу и др. в 1956 году, в котором 78% из 203 проанализированных пациентов с NF1 имели по крайней мере шесть пятен от кофе с молоком размером более 15 мм (11).
Это количество больше, чем указано для населения в целом. В ретроспективном обзоре младенцев с родимыми пятнами Mihm и соавт. показали, что у 1,8% афроамериканских младенцев будет три или более пятен цвета кофе с молоком, в то время как у кавказских младенцев редко бывает два или более (12).
Гистологически эти высыпания представляют гиперпигментацию базального эпидермиса с присутствием макромеланосом (9).
Хотя возможности для злокачественной трансформации нет, иногда требуется косметическое лечение.
Отчеты о случаях показывают, что у некоторых пациентов может быть хороший ответ на дерматологическую лазерную терапию для депигментации; однако для получения ответа может потребоваться несколько процедур, и у части пациентов рецидив пигментации произойдет в течение 6 месяцев (13).
Пигментные пятна в подмышечной впадине и паху являются еще одним определяющим симптомом NF1. Только примерно 40% пациентов будут иметь пигментные пятна в младенчестве, а у 90% пациентов с NF1 они появятся к 7 годам (1).
Меланоцитарные узелки также могут встречаться в радужной оболочке у пациентов с NF1. Эти небольшие, часто множественные гамартоматозные поражения, известные как узелки Лиша, встречаются у 93% взрослых с NF1. Они протекают бессимптомно (14).
Глиомы. Пациенты с NF1 подвергаются повышенному риску развития глиом низкой и высокой степени злокачественности. Наиболее часто встречающейся глиомой в условиях NF1 является глиома зрительного нерва низкой степени злокачественности.
Эти глиомы зрительного нерва встречаются примерно у 15% пациентов с NF1 и обычно появляются в возрасте 7 лет (2).
Как правило, эти опухоли являются пилоцитарными астроцитомами I степени по классификации Всемирной организации здравоохранения и гистологически эквивалентны пилоцитарным астроцитомам общей популяции с двухфазным ростом, волосоподобными отростками и волокнами Розенталя.
Учитывая вялотекущий характер роста этих опухолей, лечение обычно состоит из наблюдения. Когда острота зрения снижается, может быть применена химиотерапия (2).
Расположение этих опухолей не позволяет проводить хирургическое вмешательство, и у пациентов с NF1 лучевой терапии избегают из-за повышенного риска развития злокачественных новообразований, вызванных радиацией (15).
Глиомы ствола головного мозга являются второй по частоте глиомой NF1, и опять же, эти опухоли обычно являются пилоцитарными астроцитомами.
По тем же причинам, что и при глиомах зрительного нерва, химиотерапия является единственным доступным методом лечения с целью уменьшения симптомов и повышения длительности выживания.
Наконец, пациенты с NF1 имеют пятикратный риск развития злокачественных глиом, особенно глиобластом, по сравнению с общей популяцией со средней выживаемостью около 1 года.
Скелетно-мышечные нарушения. У детей с NF1 значительно повышен риск развития рабдомиосаркомы, что примерно в 20 раз выше, чем у населения в целом (2).
Эти рабдомиосаркомы могут возникать в любом месте; однако систематический обзор показал преобладание локализации в тканях мочевого пузыря и предстательной железы (16). Протоколы лечения, применяемые при несиндромных рабдомиосаркомах, также могут быть применены к пациентам с NF1.
У пациентов с NF1 часто отмечаются различные аномалии скелета, включая остеопению, сколиоз или врожденную дисплазию большеберцовой кости. Кроме того, многие пациенты с NF1 невысокого роста, хотя пропорции их тела остаются нормальными (17).
Механистическая связь NF1 и деформаций скелета в значительной степени неизвестна; однако сообщалось, что у пациентов с NF1 низкая минеральная плотность костей и низкая концентрация витамина D (2).
Исследования показали, что риск переломов у детей, страдающих NF1, увеличивается в три раза, а у взрослых-в пять раз (17, 18). Повторные переломы у этих пациентов могут привести к псевдоартрозу.
Желудочно-кишечные проявления. Желудочно-кишечный тракт может быть поражен нейрофибромами и злокачественными опухолями оболочек периферических нервов, аналогично другим участкам тела; однако также существуют различные другие злокачественные новообразования желудочно-кишечного тракта, связанные с нейрофиброматозом.
Что касается нейрофибром, желудочно-кишечный тракт может быть поражён очаговыми нейрофибромами или диффузной нейрофиброматозной пролиферацией, локализованной в собственной пластинке. В обоих случаях ганглиозные клетки могут пролиферировать без клинического значения.
Стромальные опухоли желудочно-кишечного тракта (GIST) не являются редким явлением у пациентов с NF1 и встречаются до 25% случаев (19). В отличие от GIST в общей популяции, GIST, ассоциированные с NF1, редко сочетаются с мутациями в KIT или PDGFRA.
В большинстве случаев GIST протекает бессимптомно и доброкачественно. Гистологически GIST имеет сходства с несиндромными опухолями, состоящие из веретенообразных клеток и скейноидных волокон.
Эндокринные опухоли желудочно-кишечного тракта также наблюдаются у пациентов с NF1, и они имеют склонность к локализации в периампулярной области.
Наиболее распространенной эндокринной опухолью, о которой сообщается, является соматостатинома; однако в этой ситуации также были описаны гастринома, инсулинома, карциноиды и параганглиомы (19).
Другие злокачественные новообразования. Пациенты с NF1 также имеют предрасположенность к злокачественным новообразованиям вне тканей нервной системы.
У детей с NF1 в семь раз повышен риск злокачественных новообразований кроветворной системы, особенно миелоидного лейкоза, по сравнению с их сверстниками. Лечение и прогноз аналогичны для общей популяции.
Пациенты с NF1 также подвержены повышенному риску развития рака молочной железы, особенно у женщин в возрасте до 50 лет.
Опять же, лечение одинаково для обеих групп пациентов. Наконец, хотя это редкое явление, феохромоцитомы надпочечников наблюдаются чаще у пациентов с NF1, чем в общей популяции, с зарегистрированной частотой до 5% по сравнению с менее чем 1% (2).
Проявление феохромоцитомы часто включает покраснение, учащенное сердцебиение и гипертонию, и хирургическое вмешательство часто является основным лечебным мероприятием.
Нейрофиброматоз 2 типа
Введение. NF2, также известный как двусторонний акустический нейрофиброматоз или центральный нейрофиброматоз, представляет собой наследственный опухолевый синдром, характеризующийся преимущественно развитием шванном, наряду с менингиомами, эпендимомами и аномалиями зрения.
Несмотря на название, нейрофибромы встречаются относительно редко.
NF2 наследуется по аутосомно-доминантному типу с предполагаемой частотой 1 на 25 000, распространенностью 1 на 60 000 и пенетрантностью приблизительно 0,95 (20).
Пациенты обычно находятся в возрасте около 20 лет, и прогностические аспекты включают возраст на момент постановки диагноза, фазу развития менингиомы и доступ к специализированным медицинским центрам (21).
Заболевание вызвано мутацией в гене NF2 на 22-й хромосоме, который кодирует белок мерлин. Более половины случаев вызваны мутациями гена de novo у пациентов без семейного анамнеза заболевания.
Диагностические критерии. Двусторонние шванномы верхней вестибулярной ветви восьмого черепного нерва (вестибулярная шваннома или акустическая неврома) являются патогномоничными для NF2.
Однако, поскольку у 41% пациентов, у которых в конечном итоге было доказано наличие NF2, на начальном этапе не было двусторонних вестибулярных шванном, было создано несколько диагностических стандартов для NF2.
К ним относятся широко признанные критерии Манчестера, а также дополнительные критерии NIH, приведенные в таблице 2. Бейзер и соавт. недавно предложили систему оценки для замены критериев Манчестера с якобы повышенной чувствительностью при сохранении 100% специфичности (21, 22).